
Zuker's algorithm
The Zuker algorithm calculates the optimal secondary structure of an RNA sequence with the minimum free energy under a given thermodynamic model. So it is an algorithm for RNA structure prediction. The algorithm uses the dynamic programming method and was published in 1981 . The RNA structure prediction program mfold implements a modified version of this algorithm.
idea

A given RNA sequence can fold into exponentially many different secondary structures due to the many possible combinations of base pairings. When predicting the structure, the search area must be optimized according to a certain criterion. For example, the structure with the most base pairings can be selected. However, this structure can be biologically or biochemically unrealistic, since it is z. B. contains a hairpin loop with only one unpaired base or represents an energetically very unstable structure.
A biologically more sensible criterion is the consideration of the free energy of a secondary structure. If the free energy of one structure is less than the free energy of another structure, then the first structure is more stable than the second. The free energy of various substructures of an RNA secondary structure can be measured under laboratory conditions. An example of such a data collection is. The free energy of a given RNA secondary structure of a given RNA sequence can then be approximated from this data.
The algorithm now calculates the structure with the minimum free energy for a given RNA sequence. The secondary structures that are optimal under this model are judged by experts (biologists, biochemists) to be more biologically realistic.
algorithm
The RNA sequence is denoted by, where . The minimum free energy of an optimal secondary structure is recursively calculated for all partial sequences of . The matrix stores the minimum free energy (MFE) for the partial sequence of in the cell . The matrix stores in the cell the MFE of the structure of the partial sequence , where and are a base pair.



![W [i, j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/7a0a20a8e6579edd8e691475430dfab3120830d5)
![s [i..j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/ebf63bf22305c7594ef2b27c8e639491a86fe7ac)

![V [i, j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/bc310790f650ebe3413514def0d5ce130d140ecc)
![s [i]](https://wikimedia.org/api/rest_v1/media/math/render/svg/95c8024dbbc210b378cf497974e51129a7bd62b4)
![s [j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/60c5252fdedb32516342487b03fbf6654c12a1f1)
Initialization:
![W [i, j] = 0, ji = 4](https://wikimedia.org/api/rest_v1/media/math/render/svg/f093623d7193c4363c210bf8aa00de7eb044ba4f)
![{\ displaystyle V [i, j] = 8 {,} 4 \ \ mathrm {kcal / mol}, ji = 4}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9a80ea9a19fa81583ef327fb04048adc01e3c456)
Short RNA molecules do not form a stable secondary structure.
Recursion:
![W [i, j] = \ min \ begin {Bmatrix} W [i + 1, j] \\ W [i, j-1] \\ \ min_ {i <k <j-1} \ {W [i , k] + W [k + 1, j] \} \\ V [i, j] \ end {Bmatrix}, i <j, ji> 4, 0 <i <n, 0 <j <n](https://wikimedia.org/api/rest_v1/media/math/render/svg/19685737f85b2aa1cc85c12e4a93c33e0323b25b)
The case distinction takes four cases into account. In the first or second case, the optimal structure for is composed of the optimal structure of the partial sequence or and an unpaired base to the left or right thereof. In both cases nothing changes in the MFE.
![s [i + 1, j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/ab9ef393628223ab6a462a46ae331ea6298c782b)
![s [i, j-1]](https://wikimedia.org/api/rest_v1/media/math/render/svg/c475efeeb33796f293a5dd8810a0cecd7889f505)
In the third case, the optimal structure for is formed by concatenating the optimal structures of the divided sequence. The MFE is the sum of the MFE of the substructures of the partial sequences or .
![s [i, k]](https://wikimedia.org/api/rest_v1/media/math/render/svg/1a0cbc84310badf3b731a08b00192240f786db53)
![s [k + 1, j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/3dfce3d05277342da42cb8b1a3316c9d85a4101b)
The 4th case determines the MFE for a partial sequence of if the bases and represent a base pair.
If I cannot base pair with, then will
![V [i, j] = \ inf](https://wikimedia.org/api/rest_v1/media/math/render/svg/380f914db3e6fd4cd7d6229de5179a5abe12338c)
set.
Otherwise it is calculated as follows:
![V [i, j] = \ min \ begin {Bmatrix} fh (i, j) \\ \ min_ {i <a <b <j} \ {fl (i, j, a, b) + V (a, b) \} \\ \ min_ {i + 1 <k <j-2} \ {W (i + 1, k) + W (k + 1, j-1) \} \ end {Bmatrix}, i < j, ji> 4, 0 <i <n, 0 <j <n](https://wikimedia.org/api/rest_v1/media/math/render/svg/de2dace518ac74a38d1d184bf5cea86f7190cd7b)
The function returns the free energy for a hairpin loop of the partial sequence . For example, the values for different loop sizes and base pairs can be determined experimentally under uniform conditions and are stored in an auxiliary table. The function is a substitute and supplies the free energy for an internal loop, a bulge loop or a stacking region in which the partial sequences are involved. In this case, the MFE is the sum of this construct and the MFE for the partial sequence , which must be enclosed by a base pair. The fourth case models a branch of the recursively constructed structure.


![s [i..a]](https://wikimedia.org/api/rest_v1/media/math/render/svg/e50536e6e5d4f598f34feb2957d88e05e17ea40c)
![s [b..j]](https://wikimedia.org/api/rest_v1/media/math/render/svg/b09c1f561bfc3ac481f8c7199444a5067ee40683)
![s [a, b]](https://wikimedia.org/api/rest_v1/media/math/render/svg/f2e2dd9307ce913b2b766633198d657d11c8582d)
Backtracing: After calculating the matrix recurrences, the MFE for the entire RNA sequence is stored in the cell under an optimal secondary structure. In order to determine an optimal secondary structure that the MFE has, optimization must be traced back via backtracking .
![W [0, n-1]](https://wikimedia.org/api/rest_v1/media/math/render/svg/a2f2a240599f815ecd0dbca6d816b86c163de5fc)
complexity
The two tables and are square, so the memory requirement is in . For each cell - in the case of a trivial implementation - options must be optimized, because interior loops require 2 variables. With a clever preprocessing, i.e. a pre-calculation of the values for interior loops, one can also determine this energy contribution in linear time. Alternatively, you can limit the size of a loop with a constant. So the running time of the algorithm is in .


Case distinction
The case distinction in the recursion of the recurrence of the algorithm can also be formulated more compactly if the secondary structures are mapped as Vienna strings (dot bracket strings). If a period denotes an unpaired base and a bracket denotes a paired base, then the case distinction in the recursion corresponds to the following case distinction
- ,

wherein the overall secondary structure of a substring designated and or partial structures of call.



This description is equivalent to the following graphic representation:

j unpaired
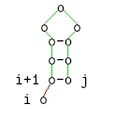
i unpaired

i and j paired

branch




Backtracing
With backtracing, several different backtracing paths can represent the same secondary structure due to the case differentiation of the Zuker algorithm. The distinction between cases is semantically ambiguous. For example, a recursion in from case 1 to case 2 to case 1 creates the same structure as the recursion from case 1 to case 1 to case 2. For another example see.
This ambiguity is not a problem when outputting an optimal secondary structure. However, if all co-optimal secondary structures are to be output or counted or all sub-optimal structures are to be output or counted up to a certain limit, then backtracing is at least difficult to implement error-free, efficiently and completely. In a standard backtracing implementation, the redundant structures are output or counted in an exponential number.
Demarcation
The algorithm uses a further table compared to the Nussinov algorithm in order to be able to evaluate a sequence of paired bases (i.e. a stacking region) differently. The Gotoh algorithm uses a similar pattern to calculate the pairwise sequence alignment for affine gap costs.
The 1978 Nussinov algorithm calculates the secondary structure of an input RNA sequence that has the maximum number of base pairs. Since these structures are not necessarily biologically meaningful, the Nussinov algorithm is not relevant in practice. In practice, among other things, RNA secondary structure prediction algorithms are used that calculate the structure with the minimum free energy or determine the structures with the smallest free energies up to a certain threshold. The use of the Zuker algorithm in the mfold implementation is still widespread, although in the meantime algorithms for secondary structure prediction also exist which undertake a detailed case distinction and whose case distinction is not ambiguous. An example of such an improved mfe algorithm is the Wuchty98 algorithm .
literature
- Michael Zuker, Patrick Stiegler: Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information . In: Nucleic Acids Research . tape 9 , no. 1 , 1981, p. 133-148 .
- Durbin et al .: Biological sequence analysis . Cambridge, 2006, ISBN 0-521-62971-3 , pp. 274-276 .
- Jens Reeder: Algorithms for RNA secondary structure analysis: prediction of pseudoknots and the consensus shapes approach . Dissertation. 2007, p. 13–15 , urn : nbn: de: hbz: 361-12764 ( uni-bielefeld.de ).
swell
- ↑ The mfold Web server. Retrieved on August 4, 2020 (official website for the mfold software).
- ↑ Michael Zuker: Free Energy and Enthalpy Tables for RNA Folding. ( Memento of April 4, 2008 in the Internet Archive ) November 3, 2000.
- ↑ Jens Reeder: Algorithms for RNA secondary structure analysis: prediction of pseudoknots and the consensus shapes approach . Dissertation. 2007, urn : nbn: de: hbz: 361-12764 .