
Fabry disease
| Classification according to ICD-10 | |
|---|---|
| E75.2 | Other sphingolipidoses (including Fabry (Anderson) disease) |
| ICD-10 online (WHO version 2019) | |

A mutation in the GLA gene, which codes for this protein, causes a reduced activity of this enzyme in patients with Fabry disease. As a result, certain fats (glycosphingolipids) cannot be broken down sufficiently and accumulate in different cells. These deposits lead to the symptoms of Fabry disease.
Fabry disease (also called Fabry disease , Fabry syndrome or Fabry-Anderson disease ) is a rare, congenital , monogenetic metabolic disorder belonging to the group of lysosomal storage diseases . The affected patients lack an enzyme (catalyst). Fabry disease is hereditary and treatable with medication.
Fabry disease is a multisystem disease that can affect a large number of organs in the body. Symptoms can vary greatly depending on the organs affected . The individually very different manifestations of the disease and its rarity make the diagnosis much more difficult ; it is usually only made correctly many years after the onset of the first symptoms. The disease primarily affects the male sex, but heterozygous (mixed-breed) women can also become ill. With them, however, the disease is usually less pronounced and only begins to become clinically relevant in middle age. The quality of life of patients with Fabry disease is often significantly impaired.
The disease has been causally treatable with enzyme replacement therapy since 2001 . The patients receive genetically engineered α-galactosidase A as an infusion throughout their life . Fabry disease is currently not curable. If left untreated, male patients reach an average age of around 50, and patients around 70 years. The main causes of early mortality are chronic kidney failure , damage to the heart, and impaired blood supply to the brain .
A mutation (genetic change) on the X chromosome (sex chromosome) greatly reduces the activity of the enzyme α-galactosidase A. It is responsible for breaking down sugary fatty substances. In the lysosomes (recycling centers of the cells), the metabolic product globotriaosylceramide (also called Gb3 or GL-3), a glycosphingolipid , can no longer be sufficiently broken down. Gb3 mainly accumulates in the cells of the inner lining of the blood vessels , the endothelial cells . As the disease progresses, these accumulations become pathological , that is, they trigger Fabry disease. Depending on the course of the disease , this can take decades.
The disease was first described in 1898 independently by the German Johannes Fabry and the English William Anderson .
Epidemiology
The disease affects all races and both sexes can get the disease. Their infrequent occurrence makes it difficult to pinpoint their frequency precisely. In the literature, prevalences of 1: 476,000 over 1: 117,000 up to 1: 40,000 are given. More recent studies based on data from newborn screenings, however, indicate a much higher incidence of Fabry disease. In northern Italy 3100 and in: a prevalence of about 1 2004-2006 Taiwan one of about 1: 1500 determined in male newborns.
If a prevalence of 1: 3100 is taken as a basis, this results in 26,450 patients with Fabry disease in Germany. Officially, around 700 people are known to be affected in Germany, in addition to a significantly higher number of unreported cases. It is assumed that in many patients the disease is not recognized during their lifetime and that premature death is attributed to other diseases. For example, in isolated cardiomyopathies , as can occur in Fabry disease patients with residual activity of α-galactosidase A. Another indication of this is that in Taiwan, 86% of the newborns that tested positive have a cryptic splice mutation of the IVS4 + 919G> A type, which was previously found mainly in Fabry disease patients with the cardiac phenotype. In these patients, Fabry's disease manifests itself mainly in a heart muscle disease (cardiomyopathy). The intronic form of this mutation is found in many Taiwanese patients with hypertrophic cardiomyopathy .
Naturally , the incidence is higher in certain subpopulations that are associated with symptoms of Fabry disease . In a study with 911 Spanish hemodialysis patients, four male and three female with changes in the GLA gene were found, which corresponds to a prevalence of 1: 182 in this subpopulation.
Genetics and molecular biology


A A 17 year old girl who has a transversion from thymine to guanine in exon 6, position 884. This nucleotide substitution changes the codon TTC, which codes for the amino acid phenylalanine , into TGC, whereby cysteine is incorporated into the α-galactosidase A gene product during translation . Consequently, in this patient's α-galactosidase A, cysteine is found in position 295 instead of phenylalanine. There is a p.Phe295Cys mutation. B A 46-year-old woman whose GLA gene in exon 1, position 125, also has a transversion from T to G. The ATG codon, which codes for methionine , therefore becomes AGG, which stands for the amino acid arginine , which is then found in position 42 of α-galactosidase A after translation. It is a p.Met42Arg mutation. C This 63-year-old patient has a GT transversion in exon 6, position 982. The nucleotide exchange turns the GGG codon into TGG and glycine becomes tryptophan , which is then found in position 328 of α-galactosidase A. The mutation is therefore called p.Gly328Trp.

Fabry disease is a disease that is due to a genetic defect (mutation) in the female sex chromosome , the X chromosome. Any father who has the condition inherits the condition to all of his daughters, while all of his sons remain healthy. If the mother carries the mutated gene, her children - regardless of gender - have a 50% risk of inheriting the disease. The mutation affects the GLA gene, which is located on the long arm of the X chromosome at the q22.1 gene locus .
The gene product is a homodimer and, like all lysosomal enzymes, is cotranslationally provided with a mannose-6-phosphate residue during the translation of the mRNA into the amino acid sequence . A portion of phosphorylated α-galactosidase A molecule is from the cells secreted and other cells via the membrane-bound mannose-6-phosphate receptor by endocytosis added. The resumption of phosphorylated α-galactosidase A via the mannose-6-phosphate receptor is the basis for enzyme replacement therapy.
Special features of the X-linked inheritance
Due to the X-linked inheritance, the disease is different in men and women. Male patients are referred to as hemizygous and female as heterozygous carriers. It used to be assumed that only men could develop Fabry disease and that heterozygous women were only carriers. This is the case with the vast majority of other X-linked hereditary diseases - such as hemophilia or muscular dystrophy of the Duchenne type . It is now known that women who have this characteristic heterozygous can get Fabry disease. Some authors therefore recommend avoiding the term X-linked recessive , as it is misleading. Instead, which is terminology X-linked inheritance recommended (Engl. X-linked inheritance ).
In heterozygous patients there is one non-mutated and one mutated X chromosome in every nucleated body cell with DNA . The X inactivation inactivates one of the two X chromosomes in each cell. This inactivation takes place in each cell independently and according to the principle of chance (so-called mosaic ). From a purely statistical point of view, 50% of the cells will therefore produce α-galactosidase A with no or reduced activity - depending on the type of mutation. The other 50% of the cells produce α-galactosidase A with normal activity (“healthy cells”). Part of the active α-galactosidase A is taken up by the "mutated cells" with the activated X chromosome, on which the defective GLA gene is located, by endocytosis, as described above. Although this enzyme transfer is sufficient to prevent the immune system from eliminating the mutated cells , it is too small to compensate for the genetic defect so that the accumulation of globotriaosylceramides is prevented. Compared to other lysosomal enzymes, the uptake of α-galactosidase A via enzyme transfer is relatively low.
The X-inactivation explains that, on average, in heterozygous women the disease becomes symptomatic much later and is less pronounced than in men. However, it is not a sufficient model to understand the wide range of different forms of the disease in women. For example, around 10% of patients require renal replacement therapy as the disease progresses , which corresponds to the “classic phenotype” in men. Other heterozygous women, however, remain largely symptom-free. The reason for this has not yet been fully clarified.
One hypothesis assumes that irregularities in the inactivation of the X chromosome play an important role in the range of variation in the heterozygous phenotype. This is known as a "crooked (engl. Skewed ) X-inactivation," in which the statistically expected 50: 50 ratio between "mutant" and "healthy" cells is significantly shifted. This shift is not caused by a growth advantage of the mutated cells. Heterozygous patients with a phenotype in which Fabry's disease is fully developed, since the mutated X chromosome is activated in more than 95% of the cells, are an indication of the crooked X inactivation. About one in 200 Fabry disease patients corresponds to this phenotype. Another indication of the crooked X-inactivation is a female heterozygous identical twin pair in which one of the twins is symptom-free and the other is clinically relevant. In a study with 28 patients, an inclined X-inactivation in the leukocytes could be found in most of the patients , but it was in no relation to the clinical manifestation of the disease or to the remaining residual enzyme activity. The authors therefore see no connection between phenotype and crooked X activation.
Mutation variants
The defects in the GLA gene caused by mutations are very heterogeneous. So far, over 500 different mutations have been recorded. These include point mutations of the missense and nonsense types , splice mutations , small deletions and insertions, and large deletions. The most common are point mutations (approx. 71%), followed by small deletions and insertions that affect less than 60 nucleotides (approx. 27%) and large deletions that affect one or more exons (approx. 2%). In most cases the mutation leads to a complete loss of enzyme activity. Some mutations that lead to changes in α-galactosidase A and are sufficiently far from the active area of the enzyme only lead to small structural changes in the enzyme, so that a certain residual activity of the enzyme is still present. Such mutations, such as p.Met72Val, p.Gln279Glu or p.Met296Ile, are characterized by a mild disease phenotype. Although the gene products have normal values for the Michaelis constant K m and the turnover rate V max , these mutated enzymes are deactivated post-translationally and then rapidly broken down. By galactose stability can these mutant enzymes are increased obviously in lymphocytes.
There is no pronounced hotspot on the GLA gene , an area particularly susceptible to mutations . What is noticeable are the increased number of DNA rearrangements in exon 7, which is obviously more susceptible to rearrangements.
pathology



Fabry's disease belongs to the group of at least 50 members of the lysosomal storage diseases and there to the subgroup of sphingolipidoses . The disease is based on a deficiency of the lysosomal enzyme α-galactosidase A. This deficit causes certain metabolic products, such as globotriaosylceramide (Gb3, Gl3, formerly also called ceramide trihexoside), to accumulate in the endothelial cells of various organ systems. The reduced activity of α-galactosidase essentially leads to an accumulation of globotriaosylceramide. In addition, digalactosaylceramide - especially in the kidneys - and globotriaosylshingosine (lyso-Gb3, lyso-Gl3) accumulate. These sphingolipids are important components of the cell membrane .
The precise relationships between reduced or even completely absent activity of α-galactosidase A and the pathological processes in the affected organs - which ultimately lead to Fabry disease - have not yet been adequately elucidated. The diversity of the affected organs suggests that secondary biochemical mechanisms in which sphingolipids play a role determine the course of the disease.
The symptoms described in the following chapter, such as progressive chronic kidney failure, are attributed in many publications to the accumulation of globotriaosylceramide in the lysosome of endothelial cells. However, a number of clinical effects, especially in enzyme replacement therapy for Fabry disease, do not fit this apparently simplified model. For example, progressive complications can be observed in some of the patients, which suggests that there is no direct correlation between Gb3 and clinical manifestation of Fabry's disease. The observation that a large proportion of female GLA mutation carriers develop symptoms that resemble those of hemizygous patients, even though these patients have considerable amounts of circulating enzyme, does not fit the simplified model . In addition, the accumulation of Gb3 in the lysosome in hemizygotic patients begins in early childhood or prenatal long before clinically relevant symptoms develop. Neither is there a correlation between the degree of disease and the plasma or urine level of Gb3 in either hemizygous or heterozygous patients.
Since the disease does not manifest itself in childhood even in patients without any activity of α-galactosidase A, it is assumed that the accumulation of Gb3 is not the immediate cause of Fabry's disease. It is currently assumed that globotriaosylsphingosine - a metabolite of globotriaosylceramide - is ultimately the cause of the pathological damage in Fabry's disease. At least in the damage to the glomeruli , which leads to renal insufficiency in Fabry disease, lyso-Gb3 plays a decisive role. Lyso-Gb3 releases TGF-β1 and the macrophage inhibitor CD74 . The pathomechanism that follows resembles that of diabetic nephropathy .
Clinical picture


Course forms and severity
A distinction is made between two types of patients and the forms of Fabry disease: the "classic" hemizygous patients in whom α-galactosidase A has no activity, and the "atypical" heterozygous patients in whom the enzyme still has residual activity. The classic course of the disease manifests itself in an early onset of symptoms, usually in several organs. In contrast, symptoms appear much later in atypical heterozygous patients. In addition, in such cases the disease can be localized, for example to the heart muscle . Male patients develop the symptoms typical of Fabry disease from childhood. In women, this is often only the case between the ages of 40 and 50. Due to the residual activity of α-galactosidase A, the symptoms are often less pronounced.
The symptoms are complex and can appear differently from person to person. The early symptoms are very important for the diagnosis. Most late symptoms, however, determine the mortality (death rate) of the patients.
The Severity Index is a key figure that determines the severity of an illness. For male Fabry disease patients, penetrance was rated as 100% and severity as 84%. For female patients, the values for penetrance are 70% and for severity 4%.
life quality
The symptoms of the disease result in a low quality of life, especially in male patients. It is similar to that of AIDS patients. In Fabry disease patients, the quality of life is on a similar level as in patients with multiple sclerosis or rheumatoid arthritis .
Fabry's disease has a significant negative impact on the psychosocial environment of those affected. Over half of the male patients are unmarried. A high proportion are unemployed. Depression is extremely common in Fabry disease patients. They are underdiagnosed or under-treated and significantly reduce the quality of life of the patient. According to a study, 46% of patients suffer from depression and 28% have depression so severe that they are clinically relevant. Values over 26 are achieved on the Hamilton scale . In contrast to the normal population, the proportion of men with severe depression (36%) is higher than that of women (22%).
Several studies recommend psychiatric and neuropsychological evaluation of patients with Fabry disease. In detail, the patients complain about physical complaints, sadness and emotional distress . Physical complaints increase under stress. Psychological tests show above-average behavior disorders, distrust, defensive attitudes, emotional agitation and a feeling of isolation. The results of these tests are largely similar to those of pain sufferers.
Early signs and symptoms


Pain
One of the first symptoms in the classic form of Fabry disease is pain in the hands and feet, the acra . These acroparesthesias appear in childhood. They are caused by damage to the thin nerve fibers ( small fiber neuropathy ) of the vegetative and peripheral somatic nervous systems . About 60 to 80% of boys and girls with the classic form are affected by this pain.
Two types of pain are described by the patients: periodically recurring attacks of pain, also known as "Fabry crises", with burning pain that radiates from the hands and feet to other parts of the body, and chronic pain, which corresponds to burning and tingling paresthesias. The Fabry crises can be triggered by fever, exercise, stress, exhaustion and rapid temperature changes. These symptoms are sometimes misinterpreted as rheumatic complaints, Raynaud's syndrome , systemic lupus erythematosus and, above all, as growing pains.
The pain usually subsides in adulthood. They occur earlier and more frequently in boys than in girls. Boys are seven years old on average and girls are nine years old. The pain has a significant negative impact on the patient's quality of life.
Gastrointestinal complaints
Digestive tract complaints are another common, mostly underestimated, early symptom of Fabry's disease. These ailments usually persist into adulthood. The patients complain of abdominal pain , mostly after eating, diarrhea , nausea and vomiting . This in turn can be the cause of anorexia (loss of appetite). These gastrointestinal symptoms are probably caused by Gb3 deposits in the autonomic ganglia (ganglia autonomica) of the intestine and the mesenteric blood vessels.
Anhidrosis
Many Fabry disease patients are unable to excrete sweat ( anhidrosis ) or can only do so to a very limited extent (hypohidrosis). The values for the skin impedance are therefore comparatively high. The anhidrosis / hypohidrosis can lead to a heat intolerance and considerable restrictions in sports activities in those affected. In a study with 714 Fabry disease patients, 53% of the male and 28% of the female subjects were diagnosed with anhidrosis. The reason for the reduced ability to secrete sweat is the accumulation of lipids within the neurons of the autonomic nervous system.
Angiokeratomas
The easiest early symptom to recognize in Fabry disease is angiokeratoma. These are benign red-purple skin changes with slight bumps. They are typically formed on the buttocks , groin , belly button, and thighs . Occasionally the mucous membranes , for example in the mouth, are also affected. In most cases, angiokeratomas are small superficial angiomas that are caused by damage to the vascular endothelium of the skin combined with vasodilation in the skin. They increase in number and size with age and can occur individually or in groups. In addition to angiomas, cases of telangiectasia and subcutaneous edema have also been reported as the cause of angiokeratomas.

Histological preparation of a skin sample taken by biopsy from a patient with Fabry disease. In the light microscope image, the typical skin lesions can be seen as small superficial angiomas.
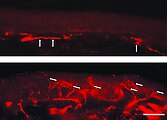
Fluorescence microscope images of a frozen section of the skin of a patient with Fabry disease.
The lack of intraepidermal nerve fibers and the presence of fibers belonging to the subepidermal nerve plexus are striking (arrows). The lower skin sample, on the other hand, comes from the patient's back. The dense innervation of the epidermis (arrows) is noticeable here.
Drawing after a microscopic examination of a carmin staining of the epidermis with an angiokeratome, watercolor from Johannes Fabry's article from 1898



Vortex Keratopathy

Characteristic corneal opacities are the most common early symptom of Fabry disease. They can be reliably diagnosed with the slit lamp and occur in almost all hemizygotic patients. This form of corneal opacity is known as cornea verticillata or vortex keratopathy . It occurs on both sides and has a characteristic cream-colored swirl-like pattern. The cloudiness does not affect visual acuity. Some drugs, such as amiodarone and chloroquine, also produce vortex keratopathy when taken for a long time.
Late symptoms
Kidney damage

Like most symptoms of Fabry disease, damage to the kidneys is progressive. It ends with terminal kidney failure and results in a significantly reduced life expectancy. In the classic clinical picture of Fabry's disease, the Gb3 accumulations in the endothelial cells of the glomerulum , in the mesangial cells , in the podocytes and in the cells of the interstitium lead to kidney damage. These cells are differentiated epithelial cells . Glycosphingolipid accumulations are also found in the epithelium of the loop of Henle and the distal tubule as well as in the endothelium and the cells of the smooth muscles of the arterioles of the kidney. The Gb3 deposits in the cytoplasm can be clearly seen in the transmission electron microscope (TEM). They are in the form of myelin structures and hit the nucleus . With increasing Gb3 accumulation, the mesangium is expanded, followed by segmental or global glomerulosclerosis with thickening of the basement membranes . Microvascular lesions and damage to the podocytes, which are important for filter performance, and the epithelial cells of the tubule are discussed as possible mechanisms.
In the classic course of the disease, kidney damage begins in the second to third decade of life . First, microalbuminuria , i.e. the excretion of small amounts of the protein albumin in the urine, can be observed, which develops into proteinuria (the excretion of larger amounts of protein in the urine). The course is similar to a diabetic nephropathy and directly contributes to the progression of Fabry nephropathy. Proteinuria becomes more and more severe with age. As the disease progresses, isosthenuria develops , that is, the kidneys completely lose their ability to concentrate or dilute. This is accompanied by changes in tubular reabsorption , secretion and excretion .
Initially, the kidney damage is masked by glomerular hyperfiltration . But once a critical number of nephrons is damaged, kidney function progressively decreases. The glomerular filtration rate , a measure of the filtration performance of the kidneys, decreases by about 12 ml / min per year if left untreated. In the third to fifth decades of life, kidney function gradually deteriorates and renal azotemia - this is the abnormal increase in nitrogen-containing metabolic products such as urea and creatinine in the blood - occurs. At this stage, fibrosis , sclerosis and tubular atrophy dominate Fabry nephropathy and ultimately lead to terminal kidney failure, which occurs in male patients in the fourth to fifth decades. About 17% of all male and 1% of all female Fabry disease patients develop end-stage renal failure and require dialysis. Half of the patients are under 53 years of age. More than half of Fabry disease patients develop nephropathy in the course of the disease . Terminal renal failure is a major contributor to morbidity and mortality . Without renal replacement therapy, uremia (urine poisoning) inevitably leads to death.
- Tissue samples from the kidney from patients with Fabry disease
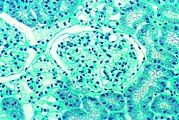
This light micrograph shows the accumulation of Gb3 in the endothelia of the glomerulum, in the mesangial cells, the cells in the interstitium and in the podocytes.
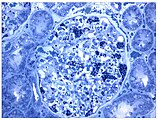
Also a light microscope image. The increased accumulation of Gb3 in the podocytes was made visible by a purple staining.

The TEM image shows the massive electron-dense (= black) accumulation of glycosphingolipids in the lysosome of the podocytes.

Also a TEM image. It shows the inclusions of glycosphingolipids of different shapes and sizes in the cells of the distal tubule.
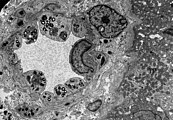
TEM image of the endothelia and smooth muscle cells of a renal artery with inclusions of glycosphingolipids





Heart damage

A Left ventricular hypertrophy (LVH) in a 51-year-old patient on dialysis with cerebrovascular involvement.
B 56-year-old patient with hypertrophic cardiomyopathy and cardiac arrhythmia, leucoaraiosis and kidney transplant.
C Late enhancement measurement after the administration of a gadolinium-containing contrast medium in a 63-year-old patient on dialysis.
About 40 to 60% of Fabry disease patients show cardiac symptoms such as left ventricular hypertrophy (LVH, thickening of the heart walls of the left ventricle ), cardiac arrhythmia, angina pectoris (attack-like pain in the chest) and dyspnoea (difficult breathing). The arrhythmia and impaired heart rate variability are caused by the sinus node , the conduction system , and a disturbance in the balance between sympathetic and parasympathetic tone . Diastolic dysfunction and left ventricular hypertrophy are major symptoms of Fabry disease. These symptoms are generally more severe for men than for women. Myocardial ischemia (circulatory disorders of the heart muscle) are the result of poor coronary blood flow.
In old age, myocardial fibroses develop progressively , which are both reversibly interstitial and irreversibly scarred (replacement fibrosis) . The irreversible cicatricial fibroses form in almost all cases first in the posterior lateral heart wall and in the midmyocardium. In end-stage patients, transmural (covering the entire thickness of the wall of the heart) cicatricial fibrosis gradually reduces cardiac function to the point of congestive heart failure . Malignant arrhythmias are responsible for most cases of cardiac death in Fabry disease patients.
The left ventricular structural changes of the heart are common in Fabry disease patients. The mostly concentric hypertrophies can be made visible using echocardiography (ultrasound examination of the heart) or cardiac magnetic resonance imaging (MRI). Since the left ventricular posterior wall of the heart becomes thinner with increasing age due to the replacement fibrosis, measuring the septum thickness - that is the thickness of the septum between the left and right halves of the heart - is particularly important. Regardless of the structural changes, the systole , the phase in which the blood is pressed out of the left and right heart chambers, appears to be largely retained when measured using conventional methods. Cardiomyopathy caused by Fabry disease is characterized by reduced contraction and relaxation of the heart muscle. Tissue Doppler (both tissue velocity imaging and strain rate imaging ) can quantify cardiac muscle function. With this method, cardiomyopathy can be diagnosed even before left ventricular hypertrophy develops.
- Echocardiographs of patients with Fabry disease

Parasternal long axis: Clearly visible left ventricular hypertrophy with increased septal thickness.

Parasternal short axis: The image also shows left ventricular hypertrophy.

Tissue Doppler echocardiography of the mitral annulus (mitral ring) with almost normal systolic function



In many Fabry disease patients, the right ventricle is also hypertrophic ( right ventricular hypertrophy , RVH). The ventricle has a normal size and the systole is also normal; the diastolic function is massively reduced. Two thirds of patients with LVH also show the symptom of RVH. Right ventricular hypertrophy is a likely cause that patients with good left ventricular heart function have poor physical endurance and suffer from organomegaly (enlarged organs) and lymphedema .
Due to the damage to the heart function, the electrocardiograms (ECG) of adult Fabry disease patients with the classic form of the disease show characteristic changes.
- Electrocardiograms (EKG) from patients with Fabry disease
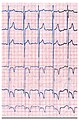
The ECG of a Fabry disease patient shows left ventricular hypertrophy with an increased Sokolow-Lyon index , reduced ST segment and negative T waves in the left ECG leads .

A 24-hour EKG is often done before and after enzyme replacement therapy if the patient reports irregular heartbeat or palpitations.


Cerebrovascular damage
The early symptoms of peripheral neuropathies , mostly occurring in adolescence, are often followed in adulthood by cerebrovascular diseases and autonomic dysfunctions (diseases or functional disorders of the autonomic nervous system). Some of the particularly unfavorable neurological characteristics of Fabry disease are caused by cerebral, multi-site (multifocal) circulatory disorders in the small blood vessels. The cerebrovascular changes can lead to a variety of different signs and symptoms. The spectrum ranges from headaches and dizziness, to transient ischemic attacks and ischemic strokes , to vascular dementia .
The prevalence of cerebral infarction is around 6.9% in male patients and 4.3% in females. It is much higher than in the general population. The mean age at first stroke is around 39 for men with Fabry disease and 46 for women. It is not uncommon for a cerebral infarction to be the first manifestation of Fabry's disease. In most cases, the cerebral infarction is triggered by small blood vessels. In addition, dolichoectasias (syn. Dilated arteriopathies , arterial widening ) of the vertebrobasilar circulation have been described as a trigger. The formation of thrombi is possibly promoted by an increased adhesion of neutrophils and monocytes to the endothelia or by a locally increased blood flow ( hyperperfusion ). The serum level of the enzyme myeloperoxidase is a biomarker of the risk of a vasculopathic incident in men with Fabry disease.
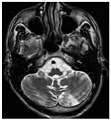
Axial MRI of the brain of a 27 year old Fabry disease patient with ischemic stroke shows the stroke in the left cerebellar hemisphere. Otherwise the patient had no symptoms of the disease.
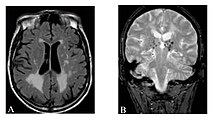
White matter hyperintensities , lacunar cerebral infarctions and microbleeding.
A) Axial MRI reveals multiple white matter lesions in the cerebral hemisphere of a 53 year old male patient with a Fazekas score of 3.
B) Lacunae and microbleeding in the same patient.
The T 1 -weighted sagittal (A) and axial (B) MRIs show a symmetrically high signal in the thalamus (the so-called pulvinar sign ) of a 66-year-old male patient. (C) and (D), also T 1 -weighted, show the Pulvinar sign in a 42-year-old patient.

The time-of-flight magnetic resonance angiographies of four Fabry disease patients show dilated (ecstatic) blood vessels (dolichoectasia of the vertebrobasilar circulation).




Further late effects



The damage caused to the kidneys, heart and brain is the major contributor to the mortality of Fabry disease. Other long-term effects are clinically relevant, but make little or no contribution to the mortality of the disease. For example, damage to the auditory and balance organs are widespread. 80% of male and 77% of female patients have a progressive loss of balance. The function of the organ of equilibrium can be checked using a head impulse test . Progressive hearing loss and sudden deafness are extremely common in hemizygotic patients with a classic course of the disease . The accumulation of α-galactosidase A can also lead to tinnitus and vertigo .
The airways are also affected by the disease in many Fabry disease patients. Difficulty breathing ( dyspnoea ), chronic cough , as well as whining and wheezing are common in both sexes. According to a study, 61% of male and 26% of female patients have airway obstruction .
Changes to the skeleton, which essentially affect bone density, are also a common late symptom of Fabry's disease. In one study, 88% of patients with an average age of 31 years and a classic course of the disease were diagnosed with either osteopenia or the advanced stage, osteoporosis , using dual X-ray absorptiometry (DXA) . In a subsequent larger study, osteopenia was found in about 50% of Fabry disease patients. The reduced bone density can lead to spontaneous fractures .
diagnosis
Importance of Early Diagnosis
Diagnosing Fabry disease as early as possible is important for several reasons. On the one hand, since 2001 there has been the option of treating the disease causally. This can significantly improve the quality of life of those affected and at least reduce or delay organ damage. On the other hand, the genetic predisposition for the disease can be recognized in family members before the first symptoms appear. In such cases, monitoring of disease development and early therapy is possible before the disease becomes symptomatic.
Misdiagnosis
Due to the rarity of the disease, Fabry disease is misdiagnosed by most pediatricians and internists and treated accordingly. A 2010 study analyzed the medical histories of 45 patients with Fabry disease. Most patients complained of neuropathic pain as the first symptom of the disease in their youth - in most cases it was misdiagnosed as " rheumatic fever " . Seven patients were treated with penicillin for years . Ten patients with abdominal pain were diagnosed with food poisoning or "unspecific pain". The initial symptom anhidrosis could not be assigned to any cause and angiokeratomas were interpreted as petechiae . It took an average of 19.7 years for the correct diagnosis of Fabry disease to be made. In a previous study with 366 patients, this time difference was 13.7 years for men and 16.3 years for women. A British study from 2001 found a mean age of 22 years in male patients for the initial diagnosis, which was made an average of 8 years after the first symptoms.
In the long period between the first symptoms and a correct diagnosis, many patients have had a long and frustrating odyssey from doctor to doctor. In most cases the correct diagnosis is made by chance at an ophthalmologist via the cornea verticillata (vortex keratopathy) or at the dermatologist via the angiokeratoma.
Correct diagnosis
In the classic form of Fabry's disease, the clinical picture can make important contributions to an early, correct diagnosis; especially angiokeratomas and vortex keratopathy. In male patients, the determination of the activity of α-galactosidase A, from plasma or from leukocytes, with an enzyme assay provides diagnostic reliability . The determination from the plasma can sometimes lead to a misdiagnosis of Fabry's disease, which is why it is recommended to check the result with the activity determination via the leukocytes. This method is often insufficient in female patients. It fails in over 30% of Fabry disease patients because the residual enzyme activity is too high for the test. Therefore, all patients suspected of having Fabry disease should be diagnosed using genotyping.
The gene is sequenced and matched with known GLA mutations. In addition, the determination of the biomarker Lyso-Gb3 is useful. If the enzyme activity is unclear, a lyso-Gb3 value higher than 1.3 nmol / l can indicate Fabry disease in people with unspecific Fabry disease symptoms (LVH or CKD, etc.). For the measurement of enzyme activity and lyso-Gb3 as well as for genetic analysis, a dry blood test (Dried Blood Spot, DBS) that can be easily integrated into everyday practice is available: For this purpose, a few drops of blood are placed on a dry blood card. After they have dried, the card is mailed to a specialized laboratory. There the blood is removed from the filter card and processed for further tests. Lyso-GL-3 is a good marker to reliably rule out classic Fabry disease. People with an uncertain GLA meaning who show unspecific Fabry disease symptoms (LVH or CKD etc.) and no characteristic, phenotypic or biochemical features of classic Fabry disease should be treated with elevated Lyso GL-3 values> 1.3 nmol / l think of Fabry's disease.
The plasma and urine levels of Gb3 can in principle also be used to make a diagnosis. The urine values are more reliable than the plasma values for male and female patients; however, some patients with the late form of Fabry disease or with certain forms of mutation (e.g. p.Asn215Ser) have normal Gb3 concentrations in the urine.
The Cobb syndrome must be differentiated from a differential diagnosis .
Prenatal diagnosis
The diagnosis of Fabry disease is possible prenatally, i.e. prenatally . This may include biochemical or molecular prenatal diagnosis are used. In the former, the activity of α-galactosidase A of chorionic villi can be measured either directly or in a cell culture . The removal by chorionic villus sampling is possible in the 10th week of pregnancy . The diagnosis of cultivated amniotic cells (cells in the amniotic fluid ), which are removed from the amniotic fluid by amniocentesis , is possible around the 14th week of pregnancy. Determining the genotype by means of DNA analysis (genetic test) is more complex . A genetic counseling is usually carried out before the prenatal diagnosis. For ethical reasons, the prenatal diagnosis of Fabry disease, especially in female fetuses, is a very controversial issue. With the availability of enzyme replacement therapy, this discussion has extended to male fetuses. Some authors recommend prenatal diagnostics only for male fetuses. The sex determination of the fetus is possible in the 9th to 11th week of pregnancy from the mother's blood.
The pre-implantation diagnosis is possible in principle and has already been carried out. So far (as of September 2011) there are no publications on this.
Newborn screening
Fabry's disease is currently not part of the newborn screenings in Germany and Austria. The premise is that screening for a specific congenital disease only makes sense if there is also a treatment option for it. With the availability of enzyme replacement therapy, this premise has become obsolete in Fabry disease. A quick and relatively inexpensive analysis of several lysosomal storage diseases can be carried out from dry blood samples ( dry blood spots , DBS) using HPLC-MS . Several large-scale studies are currently underway to verify the reliability of the procedure. Plasma-Lyso-GL-3 can easily be measured in a dry blood test and is an effective and specific biomarker for screening newborns with suspected Fabry disease before the first symptoms become visible.
therapy

The development of effective treatment methods, especially active ingredients, is extremely difficult in Fabry disease - as in all lysosomal storage diseases. On the one hand, because of the low incidence, there are only very few patients for clinical studies to be carried out and, on the other hand, the requirements with regard to drug safety are very high when taken over long periods of time. The medication should be taken for life and ideally before the onset of symptoms, i.e. by largely healthy patients. Due to the rarity of the disease, the market for a drug that has been developed is extremely small. The high development costs customary in the pharmaceutical industry are thus distributed over a small number of patients, which results in very high treatment costs per patient.
Until 2001, Fabry disease patients could only be treated symptomatically or palliatively . Up until this point, treatment consisted essentially of avoiding pain-inducing stimuli, such as stress, physical exertion, heat, sunlight and sharp changes in temperature. There were high-dose analgesics for the pain . Anhidrosis was counteracted by increasing the intake of fluids in hot weather and avoiding physical strain. A low-fat diet and medication were used to relieve gastrointestinal complaints. Diets for the kidneys were prescribed for mild proteinuria. Anticoagulant drugs have been prescribed to prevent strokes. Terminal kidney failure was treated - as it is today - with kidney replacement therapy (dialysis or kidney transplantation).
Enzyme replacement therapy
Enzyme replacement therapy (ERT) is currently (as of September 2011) the only option for the causal treatment ( causal therapy ) of Fabry's disease. For Fabry disease patients in the European Union, there are two active substances used to treat the disease: agalsidase alfa and agalsidase beta. Both are biotechnologically produced variants of α-galactosidase A. Agalsidase beta has been available for US patients since April 2003. Agalsidase alfa has not yet been approved in the United States (as of September 2011). In addition to the 27 EU countries, it is approved in a total of 45 countries, including Canada, Japan, Brazil and China (as of May 2011).
Both drugs are genetically engineered. While in agalsidase alfa a human fibroblast cell line producing the enzyme, are used in agalsidase beta ovary of Chinese hamster ( CHO cells ) were used. Agalsidase beta is a chimeric protein . The two enzymes are identical in their amino acid sequence, but differ slightly in the type of glycosylation due to the different form of protein expression during production . The main differences are in the proportion of sialic acids and mannose-6-phosphate. Agalsidase beta has a higher proportion of completely sialylated oligosaccharides and a higher degree of phosphorylation. In in-vitro tests, agalsidase beta showed increased binding to the mannose-6-phosphate receptor and a higher uptake in Fabry fibroblasts. In contrast, no functional difference between the two enzymes can be identified in vivo . They are also indistinguishable in terms of the degree of antibody cross-reactivity . Both enzymes must be administered intravenously. Enzymes that are supposed to have a systemic effect are generally not available orally, as they are largely broken down into their components (amino acids) in the intestine. Agalsidase alfa is infused at a dose of 0.2 mg / kg body weight every two weeks over a period of 40 minutes . Agalsidase beta is administered in the same rhythm. However, the dosage is 1 mg / kg body weight and the infusion time is initially four hours. This infusion time can then be reduced to 90 minutes if tolerated well.
effectiveness

Due to the slow progression of Fabry's disease over decades, as well as its rarity, there are so far only few reliable data on the long-term success of treatment. In a comparative study over a period of 24 months, no difference between agalsidase alfa and agalsidase beta could be found in the main measurable disease parameters. Especially in the endothelial cells, enzyme replacement therapy can significantly reduce the accumulation of Gb3 in the lysosome. This is of great importance for the treatment of the primary morbidity factors (kidney failure, cardiac and cerebrovascular diseases). However, the effectiveness of the therapy in patients with advanced symptoms is rather modest. Treatment as early as possible is therefore particularly important for the success of the therapy. The breakdown of Gb3 in enzyme replacement therapy depends on the cell type. In the kidneys, Gb3 is broken down - in addition to the vascular endothelia - also in the mesangial cells of the glomerulum and cells in the interstitium of the renal cortex. The Gb3 in the smooth muscles of the arterioles and small arteries, the podocytes and in the epithelium of the distal tubule is reduced significantly worse.
In clinical studies, agalsidase alfa was shown to reduce pain and left ventricular hypertrophy. The kidney function was stabilized, the hearing and the ability to perspire were improved. Overall, the quality of life has increased significantly. In patients with chronic kidney failure, the progression to terminal kidney failure could be delayed. During treatment with agalsidase beta, a depletion of Gb3 was demonstrated in various cells. The re-accumulation of Gb3 does not occur either. The administration of agalsidase beta significantly reduces the risk of a serious clinical event (e.g. myocardial infarction, terminal kidney failure, or death).
Side effects
About half of all patients have mild to moderate infusion-related reactions that peak between the fifth and eighth infusions. In addition, some patients show fever and chills . These side effects are short-lived, non-serious, and can be managed conservatively. After three to five years of treatment, only 10 to 20% of patients show infusion-related reactions, and infusion tolerance evidently develops. The exact cause of the infusion-related reactions is still unknown. A specific IgG antibody formation on the infused enzyme is suspected . IgG seroconversion was seen in 24% of patients receiving agalsidase alfa and 88% of patients receiving agalsidase beta . The formation of antibodies can occur in patients who have no residual activity of α-galactosidase. For your immune system , α-galactosidase is “new” and “strange”. The formation of antibodies against the two agalsidase preparations immediately reduces their effectiveness. A multicentre study showed, however, that an "overspray" of antibodies by corresponding amounts of enzyme results in continued low levels of the disease marker lyso-Gb3.
Competitive situation and production problems
In 2015, Shire Pharmaceuticals had Replagal sales of $ 326 million and Genzyme sales of Fabrazyme were $ 188 million. Competition between the two companies is fierce in the European market, while only Fabrazyme is approved in the United States . Genzyme lost market share in Europe because patients were switched from the full dose of Fabrazyme to lower doses or Replagal as part of a delivery bottleneck . When this delivery bottleneck with Fabrazyme arose, a large part of the American goods were allocated for export to Europe . As a result, US patients had either a lower dose or no drug available for treatment at all, while in Europe around 400 patients were adequately supplied with the full dose.
The delivery problems first arose in 2009 due to contamination with imported caliciviruses (type: Vesivirus 2117) in the world's only bioreactors for the production of Fabrazyme in Allston ( Massachusetts ). Although the virus is harmless to humans, it had a significant impact on the production yield. The quality of the product, however, was not affected. The production plant was also shut down for a long time due to disinfection measures. Genzyme was fined $ 175 million.
In August 2010, three US patients with Fabry disease petitioned the National Institutes of Health (NIH) for Genzyme to allow other companies to provide enzyme replacement therapy based on March-in Rights . The March-in Rights are special rights of intervention by the state that exist when research and development have been financed by the state. The Bayh – Dole Act of 1980 allows universities, among others, to market the results of government-funded research themselves. In turn, the Bayh-Dole Act allows funding government agencies to override exclusive rights to intellectual property (in this case, patents), for example when people are at risk. Agalsidase beta was developed at the Mount Sinai School of Medicine with NIH funds and is protected by two patents (US 5,356,804 and US 5,580,757). Genzyme is the exclusive licensee of both patents. The patient's petition was rejected by the NIH in December 2010. The main reason given by the authority is that further licensing to third parties would not solve the problems cited by the applicants - essentially the acute supply bottleneck. It would take several years from each license award to the availability of the drug from another manufacturer, since complex clinical studies and approval processes are necessary. In addition, Genzyme promise to achieve full production capacity again in the first half of 2010.
Fabrazyme's production problems persisted in September 2011. In the first quarter of 2012, production began at a new facility in Framingham, Massachusetts , and the facility was approved by the FDA. 100 US patients are currently (as of September 2011) receiving the competing product Replagal as part of a clinical study approved by the FDA .
After US regulators gave the go-ahead to manufacture Fabrazyme at the Framingham, Massachusetts site, Sanofi Genzyme Corp. began manufacturing. with delivery. The FDA and EMA previously approved the Framingham facility for the production of Fabrazyme in January 2012.
In March 2012, it was announced that Shire Pharmaceuticals had withdrawn Replagal's US approval. Despite a recommendation made by the US FDA and despite a Fast Track status granted by the FDA , it was not approved for the US market.
The Mount Sinai School of Medicine patents 5,356,804 expired on September 27, 2015 for the US market and in August 2016 for the European market.
Therapy costs
Treatment costs in Germany are around € 250,000 per patient per year, regardless of whether agalsidase alpha or beta is used. An ampoule with 3.5 mg agalsidase alfa, for a dosage of 0.2 mg per kg body weight, costs € 1,685 in France. For an ampoule with 35 mg agalsidase beta, with a dosage of 1 mg per kg body weight, 3370 € are charged. This results in identical annual treatment costs per patient, which in France are € 161,781 for a 70 kg patient (as of 2009). Since enzyme replacement therapy is currently the only causal therapeutic option for Fabry's disease, the costs for it are reimbursed by the statutory health insurances in Germany and are not offset against the prescription budget of the prescribing doctor.
Accompanying therapies
Enzyme replacement therapy reduces neuropathic pain quite successfully, especially in young patients. However, accompanying pain therapy is still indicated in many cases. For the treatment of neuropathic pain in Fabry disease, carbamazepine , possibly in combination with pregabalin, is recommended as the first choice . Opioids can also be used for intolerable pain crises . For gastrointestinal complaints, drugs such as metoclopramide are recommended, which counteract movement disorders in the upper gastrointestinal tract (motility disorders).
If there is increased protein excretion in the urine as an indication of kidney damage , the progression of kidney damage can be delayed by additional treatment with ACE inhibitors or AT1 antagonists , two related classes of antihypertensive drugs. If the heart is already damaged, ACE inhibitors can lower the arterial vascular resistance and thus the arterial blood pressure. This also reduces the preload and afterload of the heart in the case of myocardial insufficiency and increases the cardiac output . The heart rate and the oxygen demand of the myocardium can be reduced by using beta blockers . Cardiac arrhythmias can be corrected with amiodarone , for example . There are also surgical measures, such as the implantation of a pacemaker , a coronary stent , an artificial heart valve or a coronary artery bypass .
Possible future therapy options
In many cases, enzyme replacement therapy can stop or at least reduce the progression of Fabry disease. The treatment is purely palliative , which means that a complete healing is not possible with it. In addition, not all patients respond optimally to this form of treatment. The dosage form is determined for intravenous administration . Oral ingestion, which is generally preferred by patients, is not possible. During absorption in the gastrointestinal tract, α-galactosidase A is broken down into ineffective fragments. One focus of development is therefore on orally available drugs that are at least equivalent to enzyme replacement therapy in terms of their effectiveness or that can complement them therapeutically.
The enzyme replacement therapy itself is also being further developed. A possible future drug is a modified form of the enzyme α-N-acetylgalactosaminidase (NAGA). α-N-acetylgalactosaminidase has a structure very similar to α-galactosidase A. In the active pocket they only differ in two amino acid positions. Through targeted changes ( protein engineering ) in the active pocket of this human enzyme, it is able to take up Gb3 as a substrate and break it down like α-galactosidase A. The great advantage of the modified α-N-acetylgalactosaminidase is that it is not immunogenic in Fabry disease patients . Your immune system “knows” α-N-acetylgalactosaminidase.
Chaperone Therapy

Most of the mutations on the GLA gene are of the missense type . The gene products include a large number of enzyme variants which, in their purified form, are comparable in their activity to the wild type of α-galactosidase A, but have a lower thermal and pH stability. These mutant variants of the α-galactosidase A is - like all newly synthesized proteins - in the endoplasmic reticulum (ER) of a "quality control" chromatography ( Protein Quality Control , protein quality control machinery ). It ensures that only correctly folded and modified proteins reach their destination. Proteins that fail this quality control are transported from the endoplasmic reticulum into the cytosol and broken down in the proteasome . This process is called ER-associated protein degradation (ERAD, Endoplasmic Reticulum Associated Protein Degradation ). The incorrectly folded variants of α-galactosidase A due to the mutation are sorted out and broken down as part of quality control.
However, this process offers an opportunity for therapeutic intervention. With the help of pharmacological chaperones or chemical chaperones , the misfolding of the mutated enzymes can be corrected. The pharmacological chaperones are small molecules ( small molecules ) as folding template serve the enzyme. This shifts the folding dynamics of the protein towards the correct conformation and stabilizes it. With the correct tertiary structure , the quality control in the ER is "passed". The stable chaperone-protein complex is transferred through vesicles of the endoplasmic reticulum into the Golgi apparatus and then into the lysosome. There the pharmacological chaperone is replaced by the natural substrate (Gb3). The dissociation of the chaperone-protein complex is favored by the high concentration of Gb3 and the low pH value in the lysosome.
The imino sugar 1-Deoxygalactonojirimycin (DGJ), international non- proprietary name Migalastat , is an example of a pharmacological chaperone. It is an analogue of the terminal galactose of Gb3 and a reversible inhibitor of α-galactosidase A. In a large number of preclinical experiments it has been shown that migalastat is able to increase the activity of mutated variants of α-galactosidase A. This made it possible to significantly reduce the accumulation of Gb3 in Fabry mice, for example. As a small molecule, Migalastat has a very broad biodistribution in the organism and can, for example, reach the central nervous system and cross the blood-brain barrier . In addition, it is available orally.
Migalastat has been approved in the European Union since 2016. So far it is still unclear what proportion of Fabry disease patients could be treated with this form of therapy in the future, since not all of the more than 500 mutation forms of α-galactosidase A known to date can be activated or correctly developed with pharmacological chaperones. In a study of 299 forms of mutation, 40 of them are classified as potentially treatable with pharmacological chaperones. The chances of success are good, especially with the mutations that cause a non-classical course of the disease. The analysis of the genotype will be obligatory before a possible start of therapy.
Substrate reduction therapy

While enzyme replacement therapy for Fabry disease attempts to replace the missing or defective α-galactosidase by administering an effective, artificially produced enzyme in order to be able to break down the substrate Gb3 in the cells, substrate reduction therapy (SRT) takes a different approach. Here one tries to reduce the substrate and thereby prevent the accumulation of Gb3 in the cells. This is possible, for example, by inhibiting the enzyme glucosylceramide synthase (GCS, also called ceramide glucosyltransferase ). GCS catalyzes the first step in the synthesis of glycosphingolipids and thereby the synthesis of subsequent molecules, including Gb3. The basic effectiveness of this therapeutic approach was demonstrated in the mouse model organism. The SRT could become a future complementary treatment option to the enzyme replacement therapy in Fabry disease. An example of a glycosyl transferase inhibitor is miglustat . This drug is approved for the treatment of two lysosomal storage diseases: for Niemann-Pick disease type C and Gaucher disease type 1. In the latter, however, only in patients for whom ERT is not a treatment option. Miglustat is not approved for the treatment of Fabry's disease. The side effect profile of miglustat with diarrhea and peripheral neuropathy is unfavorable for Fabry's disease and could further exacerbate the symptoms of the disease. In the case of Fabry disease, in which the majority of patients show no residual activity of the mutated enzyme, SRT is rather unlikely as monotherapy . Future SRT approaches are therefore more aimed at supporting enzyme replacement therapy. One potential drug is eliglustat .
Bone marrow transplant
In the model organism mouse with the gal gene switched off , it was possible to demonstrate that the activity of α-lactosidase A increases with a stem cell transplant from the bone marrow of wild-type mice . This can be achieved with reduced conditioning , for which a gene correction of around 30% is obviously sufficient. The procedure and conditioning carry a much higher risk than enzyme replacement therapy. There are no results or experiences with this form of therapy for Fabry disease.
Gene therapy
Gene therapy is a promising future treatment option with the hope that a cure can be achieved through a single treatment . DNA or RNA with the genetic code for the GLA gene would be inserted into the body cells. The therapeutic approach would be mutated gene in the causal chain → defective enzyme → defective function → deposition of Gb3 → Fabry disease one stage earlier than in enzyme replacement therapy. In preclinical experiments on Fabry mice , promising results were obtained with various vectors , especially viral vectors . It is not yet possible to predict if and when this treatment method will be available for Fabry disease patients. In general, the high expectations in gene therapy, which existed above all at the turn of the millennium, have been significantly reduced in recent years. With some variants of the process, such as the use of retroviruses for transduction , there is a risk of inducing cancer through the inactivation of tumor suppressor genes . A successful somatic gene therapy could bring a treated Fabry disease patient a complete cure, but the defective GLA gene would still be in his germ cells . His children would therefore still have a 50% chance of getting the disease. Germ line therapies in humans that would prevent the defective gene from being passed on to offspring are prohibited in Germany and most other countries.
Animal models
Genetically modified organisms are used to develop new therapeutic options for Fabry disease and to research the disease. No large animal model for Fabry's disease has yet been discovered, which is why the a-gal-A-knockout mouse (a-gal-A - / 0 mouse or 'Fabry mouse') is essentially used as the model organism . In this knockout mouse , the gal gene is switched off by gene knockout . However, compared to the genetic defect in humans, this has little effect on the phenotype. Even at an age of 80 weeks, the mice are clinically normal, have normal blood and urine values and show the same life expectancy as the wild type . Histopathologically , the accumulation of Gb3 in the liver and kidneys can be demonstrated, which in older mice of the genotype a-Gal A - / 0 causes morphological changes in the tissue, but does not lead to any lesions. The availability of the Fabry mouse has played a major role in the development of enzyme replacement therapy for Fabry disease, especially in the preclinical phase . Α-Gal-A - / 0 mice are also used in preclinical studies on gene, chaperone and substrate reduction therapy.
forecast
With increasing age, the damage to vital organs caused by the accumulation of Gb3 increases until these organs completely lose their function. Terminal renal insufficiency and life-threatening cardiovascular or cerebrovascular complications limit the life expectancy of untreated patients to an average of around 50 and that of patients to around 70 years. Compared to the total population, this corresponds to a reduction of 20 or 15 years.
In one study, the main causes of death were kidney failure, cerebrovascular disease (for example, cerebral haemorrhage or stroke), and heart disease. The proportion of deaths from kidney failure decreased with the introduction of dialysis and transplantation. The primary cause of death in the study period from 2001 to 2007 was heart disease; 34% of men and 57% of women. The cause of the shift in the causes of death is seen in the improved clinical care of patients with end-stage renal disease.
No statistically significant data are yet available on whether and, if so, to what extent enzyme replacement therapy can increase the life expectancy of patients.

The Kaplan-Meier survival rate for male Fabry disease patients compared to the normal male population. The average life expectancy is 50 years, which corresponds to a reduction of around 20 years. The data are from 2001, when enzyme replacement therapy was not yet available.

The corresponding survival rate for female patients. The mean life expectancy is 70 years, which corresponds to a reduction of around 15 years. These data are also from 2001.


Medical history
Fabry's disease was discovered relatively late as an independent syndrome. The first publications on the disease are by Johannes Fabry and William Anderson (both in 1898). The general median life expectancy was around 30 years in 1820 and around 40 years in 1900. In comparison, the mean life expectancy of male patients with Fabry disease today is around 45 to 50 years. The disease is therefore not only extremely rare, but it was also clinically inconspicuous given the generally short life expectancy. At the end of the 18th century, dermatologists could only describe skin diseases. The significance of accompanying symptoms and the interrelationship between them was largely unknown.
The phase of empirical dermatology did not end until the 1970s with the advent of microbiological and immunological procedures.
Johannes Fabry and his patient

The German dermatologist Johannes Fabry was working at the Dortmund City Hospital at the time . In December 1898 he published the article A contribution to the knowledge of the purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae) in the archive for dermatology and syphilis .
In his article, Fabry named the skin lesions caused by angiokeratomas, Purpura papulosa haemorrhagica Hebrae . Fabry chose this name,
“... because above all we have to avoid adding a new clinical picture to the large and much controversial group of lichen; but then above all because the Hebraic term in the word 'purpura' at the same time encompasses the gender and the epithet the clinical variety and peculiarity of the nodular form and the papular rash. "
Fabry was aware of the first description. He wrote: "But first the exact medical report of the interesting and, I believe, in the literature without an analogue case."

He then described the medical history of Emil Honke, a 13-year-old boy from Langendreer . His parents noticed small nodules in the popliteal region of his knee when he was nine years old. The rash spread down the back of the thighs all the way to the trunk over the years. At twelve, the nodules appeared on the left hollow of the knee. In the first few years there were no further symptoms, but then Emil became increasingly weak and less appetite. The skin condition itself did not cause him any discomfort - no itching, no stinging, no pain and no discomfort. Both parents - in their early 40s - described Fabry as healthy. The paternal grandfather died of kidney failure at the age of 49. Fabry made the findings on April 15, 1897. He kept Emil for several days in the skin ward of the Dortmund city hospital and took tissue samples from the skin. Fabry prescribed iron drops and refrained from local treatment. Fabry described the nodules as dark blue all the way to black. Fabry described the feeling of touch when stroking the skin of the chest as “the typical feeling of a grater”, similar to, for example, Darier's disease .
Before its publication, Fabry presented the histopathological findings obtained from Emil Honke in 1897 at the meeting of doctors in the Arnsberg administrative district and in the dermatological section of the Braunschweig natural researchers' meeting.

After 17 years, on February 11, 1915, Emil Honke returned to Fabry because of his ailment. This had meanwhile completely lost sight of the case. Honke had meanwhile worked as a house painter and then as a miner for three years.
“We had now completely lost sight of this case and were of course very surprised when [the] patient presented himself again for an examination this year. It should be emphasized from the anamnesis that the skin condition did not prevent him from pursuing his profession. "
In a 1916 article on angiokeratomas, Fabry published the test result with two images of his patient. This shows the progressive spread of the angiokeratomas, especially on the back and arms, compared to the 1898 image. Fabry was able to detect albumin, i.e. proteinuria, in the urine. However, he paid no further attention to this finding. Fabry made the prognosis that no threatening development was to be expected due to the previous course of the disease. The general well-being is not disturbed and is unlikely to change in the foreseeable future. He did not expect a cure either, as the skin changes progressively increased over the years. Fabry refrained from any treatment, especially since his patient did not want it either. However, he would have liked to try a treatment with X-rays and radium .
In this publication Fabry assumed that his case of a universal angiokeratoma had no analogue in the literature: "The only possible case, Anderson's, is not an angiokeratoma, it is multiple angiomas." At this point Fabry was wrong.
In 1930, the year he died, Fabry published an article on angiokeratoma naeviforme . In it he went back to the case of his patient. Emil Honke died in 1928 at the age of 43 from an unspecified lung disease, but not from tuberculosis .
William Anderson and his patient


In April 1898, the British anatomist William Anderson published a five-page case report on a patient with an angiokeratoma (A case of 'Angeio-keratoma') in the British Journal of Dermatology .
William Anderson saw his patient (WH) for the first time in December 1897. He was 39 years old at the time and a painter by profession. His skin changes started at the age of eleven; first on the knees, then they spread to the trunk and extremities. At 17, the maximum spread was reached. Anderson recorded the spread of angiokeratoma corporis diffusum in a sketch that was part of his publication . The family history revealed similar skin changes in the mother, sister and three of four children. On the first examination, Anderson found some albumin in the patient's urine. After that, the urine was normal. After a few days in the hospital, the lesions decreased somewhat. At the request of the patient, no treatment was given.
Anderson assumed that the disease occurs in both sexes and is accompanied by chilblains or asphyxia . He said that in many cases family members could also be affected, but since no other cases of angiokeratoma corporis diffusum were known in his patient's family at the time of the anamnesis , he saw no convincing evidence that a hereditary disease was present.
WH died in 1911 of cachexia caused by tuberculous enteritis . A daughter and two grandchildren also developed Fabry's disease.
Further research into the disease
In the period that followed, Fabry's disease was initially referred to as angiokeratoma corporis diffusum . The name angiokeratoma goes back to Ernest Wyndham Cottle (? –1919), who in 1877 first described a case of angiokeratomas in a total of eight patients with ' warty growths' . Over time, the current name Fabry's disease became common , although Anderson-Fabry's disease would be more appropriate given the historical background.
Johannes Weicksel (1882–?) Was the first to recognize the characteristic changes in the retina and conjunctiva in 1925 as a symptom of Fabry's disease. In 1947, a pair of brothers was found to be involved in the progression of Fabry disease for the first time. During the autopsy, the working group around the Groningen dermatologist Maximiliaan Ruiter (1900–1974) discovered abnormal vacuoles in the blood vessels. They were the first to suspect that Fabry's disease is a systematized storage disease. In some works, Fabry's disease is therefore also referred to as Ruiter-Pompen-Weyers syndrome.
The German pathologist Karl Scriba (1907–1978) from the University Medical Center Hamburg-Eppendorf identified lipids in the vacuoles in 1950. Three years later, together with Hans Hornbostel , he was able to detect the endothelial fat deposits for the first time in a living Fabry disease patient.
In 1958, a case of Fabry disease was first described in a woman.
Charles C. Sweeley and Bernard Klionsky from the University of Pittsburgh characterized the fat deposits in the kidneys of a 28-year-old Fabry patient who had died in 1963 and identified globotriaosylceramide. They classified the disease as sphingolipidosis.
The fact that the disease has an X-linked inheritance was proven in 1965 by a research group led by John Opitz at the University of Wisconsin – Madison by means of family tree analysis . In the same year, the Japanese dermatologist Ken Hashimoto , who lives in the United States, discovered inclusion bodies in the cells in the lysosomes on TEM images of skin samples from patients with Fabry disease. He was the first to suspect that a defect in a lysosomal enzyme is the cause of the disease.
Development of enzyme replacement therapy
In 1967 a research group at the National Institute of Neurological Diseases and Blindness discovered that a deficit in ceramide trihexosidase is the genetic cause of Fabry disease. In 1970 the enzyme α-galactosidase was identified, of which the two variants A and B could be isolated from human placentas in 1978 ; a deficiency in variant A is responsible for the development of Fabry disease. With these findings in 1973, the carriers of traits - also prenatally - could be determined biochemically on the basis of the enzyme activity.
This laid the foundation for enzyme replacement therapy. Initially, the complex isolation of the enzyme from human placenta, spleen and plasma was started, and after the infusion of α-galactosidase A, a reduction in globotriaosylceramide in the blood of patients with Fabry disease was found.
The advances in genetic engineering made it possible to create genetically modified organisms . In 1996, an interdisciplinary working group under the umbrella of the National Institutes of Health succeeded in knocking out the α-Gal-A gene in the color mouse model organism , creating the first “Fabry mouse”. With the development of DNA sequencing, further advances in research into Fabry's disease have been made. Knowing the sequence of the GLA gene made it possible for the first time to localize and characterize the mutations and to produce recombinant DNA .
With the recombinant DNA introduced into bacteria, it was possible to produce α-galactosidase A as a recombinant protein and thus use it in sufficient quantities for preclinical and clinical studies. The first preclinical studies were carried out in 1996 on Fabry mice. As part of a clinical study in 1999, patients received the genetically engineered α-galactosidase A for the first time. At the beginning of August 2001, Agalsidase beta (Genzyme Corporation) and agalsidase alfa ( Transkaryotic Therapies , now Shire Pharmaceuticals) were simultaneously approved in the European Union , according to the Orphan Drug Act for the Treatment of Fabry Disease. Fabrazyme was launched on August 15, 2001, and Replagal was launched in Germany in mid-October of the same year .
literature
Reference books
- D. Elstein, G. Altarescu, M. Beck (Eds.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4 limited preview in the Google book search
- JA Barranger, MA Cabrera-Salazar (Ed.): Lysosomal storage disorders. Verlag Springer, 2007, ISBN 978-0-387-70908-6 , limited preview in the Google book search
- C. Wanner: Fabry disease: Clinic, diagnostics and therapy. Verlag Uni-Med, 2004, ISBN 3-89599-769-2 .
- A. Mehta, M. Beck, G. Sunder-Plassmann (Eds.): Fabry Disease. Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290683 (freely accessible in full)
Technical article
- B. Hoffmann, E. Mayatepek: Fabry's disease - often seen, rarely recognized . In: Dtsch Arztebl Int. 106, 2009, pp. 72-81. PMID 19623315
- B. Hoffmann: Fabry disease: recent advances in pathology, diagnosis, treatment and monitoring. In: Orphanet Journal of Rare Diseases. 4, 2009, 21. doi: 10.1186 / 1750-1172-4-21 , PMC 2768700 (free full text), PMID 19818152 (review article in Open Access )
- C. Whybra, M. Ries, C. Kampmann: Clinical manifestation of Fabry disease in children. (PDF; 2.8 MB) In: Neuropaediatrics. 2, 2005, pp. 44-48.
Non-fiction and popular science literature
- Gerald Uhlig- Romero: And yet I live. My struggle with a puzzling disease. Deutsche Verlags-Anstalt, 2009, ISBN 978-3-421-04326-9 .
- U. Schmidt: Rare hereditary disease. (PDF; 6.7 MB) In: JOGU. 186, 2003, pp. 14-15.
- M. Pawlik: Give me a place without fear! In: FAZ , June 22, 2009 (review of Gerald Uhlig's book)
- V. Hackenbroch: fat in the organ . In: Der Spiegel . No. 28 , 2006, pp. 111 ( online ).
- N. Schäufler: An orphan of medicine - The hereditary disease Fabry disease is unknown to many doctors. In: FAZ , June 12, 2002, p. 10.
Web links
- Fabry disease. In: Online Mendelian Inheritance in Man . (English)
- Fabry disease. In: Orphanet (Rare Disease Database).
- Fabry disease Pathology - Pathopic image database of the University of Basel ( PathoPic - Instructions (PDF); PDF; 2.2 MB)
- Fabry disease self-help group e. V.
- Stephanie Barbara Schatz: X-inactivation in heterozygous patients with regard to X-linked Fabry disease. (PDF) Dissertation, Ludwig Maximilians University Munich, 2012
Individual evidence
- ↑ BJ Poorthuis, RA Wevers u. a .: The frequency of lysosomal storage diseases in The Netherlands. In: Human genetics. Volume 105, Numbers 1-2, 1999 Jul-Aug, pp 151-156, PMID 10480370 .
- ↑ PJ Meikle, JJ Hopwood et al. a .: Prevalence of lysosomal storage disorders. In: JAMA. Volume 281, Number 3, January 1999, pp. 249-254, doi: 10.1001 / jama.281.3.249 , PMID 9918480 .
- ↑ a b RJ Desnick, YAIoannou, CM Eng: a-Galactosidase A deficiency: Fabry disease. In: CR Scriver , AL Beaudet, WS Sly, D. Valle (Eds.): The metabolic and molecular basis of inherited disease. 8th edition, McGraw-Hill Verlag, 2001, ISBN 0-07-913035-6 , pp. 3733-3774.
- ↑ D. Marsden, H. Levy: Newborn screening of lysosomal storage disorders. In: Clinical chemistry. Volume 56, number 7, July 2010, pp. 1071-1079, doi: 10.1373 / clinchem.2009.141622 . PMID 20489136 . (Review).
- ↑ M. Spada, S. Pagliardini et al. a .: High incidence of later-onset fabry disease revealed by newborn screening. In: American journal of human genetics. Volume 79, Number 1, July 2006, pp. 31-40, doi: 10.1086 / 504601 . PMID 16773563 . PMC 1474133 (free full text).
- ↑ a b WL Hwu, YH Chien u. a .: Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G> A (IVS4 + 919G> A). In: Human mutation. Volume 30, number 10, October 2009, pp. 1397-1405, doi: 10.1002 / humu.21074 . PMID 19621417 . PMC 2769558 (free full text).
- ↑ a b c d e f g h i j k l m n o p q r s t u v w x y z aa DP Germain: Fabry disease. In: Orphanet Journal of Rare Diseases. Volume 5, 2010, p. 30, doi: 10.1186 / 1750-1172-5-30 . PMID 21092187 . PMC 3009617 (free full text). (Review in Open Access ).
- ↑ The competence center for Fabry disease introduces itself. (PDF) ( Page no longer available , search in web archives ) Info: The link was automatically marked as defective. Please check the link according to the instructions and then remove this notice. (PDF; 595 kB) Cologne University Hospital, accessed on September 1, 2011
- ^ A b C. Vetter: Repetition: Fabry disease. (PDF) ( Page no longer available , search in web archives ) Info: The link was automatically marked as defective. Please check the link according to the instructions and then remove this notice. In: ZM. 101, No. 1A, from January 1, 2011, pp. 41-45.
- ↑ a b KF Gold, GM Pastores u. a .: Quality of life of patients with Fabry disease. In: Qual Life Res. Volume 11, Number 4, June 2002, pp. 317-327, PMID 12086117 .
- ^ HY Lin, KW Chong et al. a .: High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. ( Memento of the original from December 27, 2016 in the Internet Archive ) Info: The archive link was inserted automatically and has not yet been checked. Please check the original and archive link according to the instructions and then remove this notice. In: Circulation. Cardiovascular genetics. Volume 2, number 5, October 2009, pp. 450-456, doi: 10.1161 / CIRCGENETICS.109.862920 . PMID 20031620 .
- ↑ P. Gaspar, J. Herrera et al. a .: Frequency of Fabry disease in male and female hemodialysis patients in Spain. In: BMC medical genetics. Volume 11, 2010, p. 19, doi: 10.1186 / 1471-2350-11-19 . PMID 20122163 . PMC 2837018 (free full text).
- ↑ a b c d AP Burlina, KB Sims u. a .: Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. In: BMC neurology. Volume 11, 2011, p. 61, doi: 10.1186 / 1471-2377-11-61 . PMID 21619592 . PMC 3126707 (free full text).
- ^ A b M. Pavelka, J. Roth: Functional ultrastructure: Atlas of the biology and pathology of tissues. Verlag Springer, 2005, ISBN 3-211-83563-6 , pp. 104-105, doi : 10.1007 / 3-211-30826-1_56 restricted preview in the Google book search
- ↑ omim.org: OMIM Gene / Loci: 413 - 422 of 718 on Chromosome X. Retrieved September 1, 2011
- ^ MW King: Introduction to Fabry Disease. Status: February 13, 2011, accessed on September 1, 2011
- ↑ K. Falke: Investigations on the microstructure of the cornea verticillata in Fabry disease and comparison to the drug-induced form of the cornea verticillata by means of confocal in vivo microscopy. Dissertation, University of Rostock, 2009
- ↑ HF Willard: The sex chromosomes and X chromosome inactivation. In: CR Scriver , AL Beaudet, WS Sly, D. Valle (Eds.): The metabolic and molecular basis of inherited disease. Volume 3, 8th Edition, MacGraw-Hill, 2001, ISBN 0-07-136322-X , pp. 1191-1211.
- ↑ a b c LL Pinto, TA Vieira u. a .: Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. In: Orphanet Journal of Rare Diseases. Volume 5, 2010, p. 14, doi: 10.1186 / 1750-1172-5-14 . PMID 20509947 . PMC 2889886 (free full text). (Review in Open Access )
- ^ A b DP Germain, K. Benistan, L. Angelova: X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease. In: La Revue de médecine internal. Volume 31 Suppl 2, December 2010, pp. S209-S213, doi: 10.1016 / S0248-8663 (10) 70013-8 . PMID 21211665 .
- ↑ a b WB Dobyns, A. Filauro u. a .: Inheritance of most X-linked traits is not dominant or recessive, just X-linked. In: American journal of medical genetics. Part A. Volume 129A, number 2, August 2004, pp. 136-143, doi: 10.1002 / ajmg.a.30123 . PMID 15316978 .
- ↑ WB Dobyns: The pattern of inheritance of X-linked traits is not dominant or recessive, just X-linked. In: Acta paediatrica. Volume 95, number 451, April 2006, pp. 11-15, doi: 10.1111 / j.1651-2227.2006.tb02383.x . PMID 16720459 .
- ^ BR Migeon: X inactivation, female mosaicism, and sex differences in renal diseases. In: JASN. Volume 19, Number 11, November 2008, pp. 2052-2059, doi: 10.1681 / ASN.2008020198 . PMID 18448583 . (Review).
- ↑ M. Kobayashi, T. Ohashi et al. a .: Clinical manifestations and natural history of Japanese heterozygous females with Fabry disease. In: Journal of inherited metabolic disease. [electronic publication before printing] January 2008, doi: 10.1007 / s10545-007-0740-6 . PMID 18202903 .
- ^ BR Migeon: Females Are Mosaics: X Inactivation and Sex Differences in Disease. New York, Oxford University, 2007, ISBN 978-0-19-518812-7 .
- ↑ I. Redonnet-Vernhet, JK Ploos van Amstel u. a .: Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene. In: Journal of medical genetics. Volume 33, Number 8, August 1996, pp. 682-688, PMID 8863162 . PMC 1050704 (free full text).
- ↑ EM Maier, S. Osterrieder u. a .: Disease manifestations and X inactivation in heterozygous females with Fabry disease. In: Acta paediatrica. Volume 95, number 451, April 2006, pp. 30-38, doi: 10.1111 / j.1651-2227.2006.tb02386.x . PMID 16720462 .
- ↑ a b c A. Gal, E. Schäfer, I. Rohard: The genetic basis of Fabry disease. In: A. Mehta, M. Beck, G. Sunder-Plassmann (Eds.): Fabry Disease: Perspectives from 5 Years of FOS. Chapter 33, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290673
- ^ A. Gal: Molecular genetics of Fabry disease and Genotype-phenotype correlation. In: D. Elstein, G. Altarescu, M. Beck (Eds.): Fabry disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , pp. 3-19. limited preview in Google Book search
- ^ RO Brady : Fabry Disease - An Overview. In: D. Elstein, G. Altarescu, M. Beck (Eds.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , p. XIX limited preview in the Google book search
- ↑ a b MD Sanchez-Niño, AB Sanz u. a .: Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. In: Nephrology, dialysis, transplantation. Volume 26, Number 6, June 2011, pp. 1797-1802, doi: 10.1093 / ndt / gfq306 . PMID 20504837 .
- ↑ a b AH Futerman, G. van Meer: The cell biology of lysosomal storage disorders. In: Nature Reviews Molecular Cell Biology . Volume 5, Number 7, July 2004, pp. 554-565, doi: 10.1038 / nrm1423 . PMID 15232573 . (Review).
- ↑ M. Fuller: Sphingolipids: the nexus between Gaucher disease and insulin resistance. In: Lipids in health and disease. Volume 9, 2010, p. 113, doi: 10.1186 / 1476-511X-9-113 . PMID 20937139 . PMC 2964722 (free full text). (Review in Open Access ).
- ↑ a b c AC Vedder, GE Linthorst u. a .: Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg / kg. In: PloS one. Volume 2, number 7, 2007, p. E598, doi: 10.1371 / journal.pone.0000598 . PMID 17622343 . PMC 1913555 (free full text). ( Open access )
- ↑ KD MacDermot, A. Holmes, AH Miners: Natural history of Fabry disease in affected males and obligate carrier females. In: Journal of inherited metabolic disease. Volume 24 Suppl 2, 2001, pp. 13-14, PMID 11758673 .
- ↑ C. Whybra, C. Kampmann et al. a .: Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. In: Journal of inherited metabolic disease. Volume 24, Number 7, December 2001, pp. 715-724, PMID 11804208 .
- ↑ PB Deegan, AF Baehner a. a .: Natural history of Fabry disease in females in the Fabry Outcome Survey. In: Journal of medical genetics. Volume 43, Number 4, April 2006, pp. 347-352, doi: 10.1136 / jmg.2005.036327 . PMID 16227523 . PMC 2563231 (free full text).
- ^ S. Gupta, M. Ries u. a .: The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. In: Medicine. Volume 84, Number 5, September 2005, pp. 261-268, PMID 16148726 .
- ↑ a b c AC Vedder, GE Linthorst u. a .: The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. In: Journal of inherited metabolic disease. Volume 30, Number 1, February 2007, pp. 68-78, doi: 10.1007 / s10545-006-0484-8 . PMID 17206462 .
- ↑ AC Vedder, A. Strijland u. a .: Manifestations of Fabry disease in placental tissue. In: Journal of inherited metabolic disease. Volume 29, Number 1, February 2006, pp. 106-111, doi: 10.1007 / s10545-006-0196-0 . PMID 16601876 .
- ↑ a b E. Young, K. Mills et al. a .: Is globotriaosylceramide a useful biomarker in Fabry disease? In: Acta paediatrica. Volume 94, Number 447, March 2005, pp. 51-54, PMID 15895713 .
- ↑ S. Bekri, O. Lidove et al. a .: The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry disease: a review of the literature. In: Cardiovascular & hematological agents in medicinal chemistry. Volume 4, Number 4, October 2006, pp. 289-297, PMID 17073606 . (Review).
- ↑ JM Aerts, JE Groener u. a .: Elevated globotriaosylsphingosine is a hallmark of Fabry disease. In: PNAS . Volume 105, Number 8, February 2008, pp. 2812-2817, doi: 10.1073 / pnas.0712309105 . PMID 18287059 . PMC 2268542 (free full text).
- ↑ a b AH Miners, A. Holmes u. a .: Assessment of health-related quality-of-life in males with Anderson Fabry Disease before therapeutic intervention. In: Quality of life research. Volume 11, Number 2, March 2002, pp. 127-133, PMID 12018736 .
- ↑ a b DA Laney, DJ Gruskin u. a .: Social-adaptive and psychological functioning of patients affected by Fabry disease. In: Journal of inherited metabolic disease. January 2010, doi: 10.1007 / s10545-009-9025-6 . PMID 20087663 .
- ↑ a b AJ Grau, M. Schwaninger u. a .: A lysosomal metabolic disease with new therapeutic possibilities. In: The neurologist. Volume 74, Number 6, June 2003, pp. 489-496, doi: 10.1007 / s00115-003-1513-6 . PMID 12799787 .
- ↑ J. Sadek, R. Shellhaas et al. a .: Psychiatric findings in four female carriers of Fabry disease. In: Psychiatric genetics. Volume 14, Number 4, December 2004, pp. 199-201, PMID 15564893 .
- ↑ a b AL Cole, PJ Lee et al. a .: Depression in adults with Fabry disease: a common and under-diagnosed problem. In: Journal of inherited metabolic disease. Volume 30, Number 6, November 2007, pp. 943-951, doi: 10.1007 / s10545-007-0708-6 . PMID 17994284 .
- ↑ P. Segal, Y. Kohn et al. a .: Psychiatric and cognitive profile in Anderson-Fabry patients: a preliminary study. In: Journal of inherited metabolic disease. Volume 33, Number 4, August 2010, pp. 429-436, doi: 10.1007 / s10545-010-9133-3 . PMID 20549363 .
- ↑ TW Crosbie, W. Packman, S. Packman: Psychological aspects of patients with Fabry disease. In: Journal of inherited metabolic disease. Volume 32, Number 6, December 2009, pp. 745-753, doi: 10.1007 / s10545-009-1254-1 . PMID 19924564 .
- ↑ WJ Cable, EH Kolodny, RD Adams: Fabry disease: impaired autonomic function. In: Neurology. Volume 32, Number 5, May 1982, pp. 498-502, PMID 6803189 .
- ↑ M. Dütsch, H. Marthol u. a .: Small fiber dysfunction predominates in Fabry neuropathy. In: Journal of clinical neurophysiology. Volume 19, Number 6, December 2002, pp. 575-586, PMID 12488789 .
- ↑ B. Hoffmann, M. Beck et al. a .: Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy - a retrospective analysis from the Fabry Outcome Survey. In: The Clinical journal of pain. Volume 23, Number 6, 2007 Jul-Aug, pp. 535-542, doi: 10.1097 / AJP.0b013e318074c986 . PMID 17575495 .
- ↑ a b RJ Hopkin, J. Bissler u. a .: Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. In: Pediatric research. Volume 64, Number 5, November 2008, pp. 550-555, doi: 10.1203 / PDR.0b013e318183f132 . PMID 18596579 .
- ↑ J. Charrow: A 14-year-old boy with pain in hands and feet. In: Pediatric annals. Volume 38, Number 4, April 2009, pp. 190, 192, PMID 19455947 .
- ↑ MJ Hilz, B. Stemper, EH Kolodny: Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. In: Pain. Volume 84, Number 2-3, February 2000, pp. 361-365, PMID 10666542 .
- ↑ KJ Sheth, SL Werlin u. a .: Gastrointestinal structure and function in Fabry's disease. In: The American Journal of Gastroenterology. Volume 76, Number 3, September 1981, pp. 246-251, PMID 6274188 .
- ↑ a b B. Hoffmann, M. Schwarz u. a .: Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. In: Clinical gastroenterology and hepatology. Volume 5, number 12, December 2007, pp. 1447-1453, doi: 10.1016 / j.cgh.2007.08.012 . PMID 17919989 .
- ^ WH Kang, SI Chun, S. Lee: Generalized anhidrosis associated with Fabry's disease. In: Journal of the American Academy of Dermatology. Volume 17, Number 5 Pt 2, November 1987, pp. 883-887, PMID 3119682 .
- ↑ a b c d CH Orteu, T. Jansen u. a .: Fabry disease and the skin: data from FOS, the Fabry outcome survey. In: The British journal of dermatology. Volume 157, Number 2, August 2007, pp. 331-337, doi: 10.1111 / j.1365-2133.2007.08002.x . PMID 17573884 .
- ↑ a b SN Gupta, M. Ries u. a .: Skin-impedance in Fabry Disease: a prospective, controlled, non-randomized clinical study. In: BMC neurology. Volume 8, 2008, p. 41, doi: 10.1186 / 1471-2377-8-41 . PMID 18990229 . PMC 2585087 (free full text). ( Open access )
- ^ A b ED Shelley, WB Shelley, TW Kurczynski: Painful fingers, heat intolerance, and telangiectases of the ear: easily ignored childhood signs of Fabry disease. In: Pediatric dermatology. Volume 12, Number 3, September 1995, pp. 215-219, PMID 7501549 .
- ↑ CM Eng, DP Germain et al. a .: Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. In: Genetics in medicine. Volume 8, Number 9, September 2006, pp. 539-548, doi: 10.1097 / 01.gim.0000237866.70357.c6 . PMID 16980809 . (Review).
- ↑ M. Möhrenschlager, M. Braun-Falco u. a .: Fabry disease: recognition and management of cutaneous manifestations. In: American journal of clinical dermatology. Volume 4, Number 3, 2003, pp. 189-196, PMID 12627994 . (Review).
- ↑ D. Wattanasirichaigoon, J. Svasti et al. a .: Clinical and molecular characterization of an extended family with Fabry disease. In: Journal of the Medical Association of Thailand. Volume 89, Number 9, September 2006, pp. 1528-1535, PMID 17100396 .
- ↑ C. Orssaud, J. Dufier, D. Germain: Ocular manifestations in Fabry disease: a survey of 32 hemizygous male patients. In: Ophthalmic genetics. Volume 24, Number 3, September 2003, pp. 129-139, PMID 12868031 .
- ↑ TT Nguyen, T. Gin et al. a .: Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Center. In: Clinical & experimental ophthalmology. Volume 33, Number 2, April 2005, pp. 164-168, doi: 10.1111 / j.1442-9071.2005.00990.x . PMID 15807825 .
- ^ A. Sodi, AS Ioannidis et al. a .: Ocular manifestations of Fabry's disease: data from the Fabry Outcome Survey. In: The British journal of ophthalmology. Volume 91, Number 2, February 2007, pp. 210-214, doi: 10.1136 / bjo.2006.100602 . PMID 16973664 . PMC 1857640 (free full text).
- ↑ K. Falke, A. Büttner u. a .: The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: a confocal laser-scanning microscopy study. In: Graefe's archive for clinical and experimental ophthalmology. Volume 247, Number 4, April 2009, pp. 523-534, doi: 10.1007 / s00417-008-0962-9 . PMID 18931853 .
- ↑ A. Sessa, M. Meroni et al. a .: Renal pathological changes in Fabry disease. In: Journal of inherited metabolic disease. Volume 24 Suppl 2, 2001, pp. 66-70, PMID 11758681 . (Review).
- ↑ A. Sessa, M. Meroni et al. a .: Evolution of renal pathology in Fabry disease. In: Acta paediatrica. Volume 92, Number 443, December 2003, pp. 6-8, PMID 14989458 .
- ↑ a b AB Fogo, L. Bostad u. a .: Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). In: Nephrology, dialysis, transplantation. Volume 25, Number 7, July 2010, pp. 2168-2177, doi: 10.1093 / ndt / gfp528 . PMID 19833663 . PMC 2902894 (free full text).
- ↑ MC Gubler, G. Lenoir and a .: Early renal changes in hemizygous and heterozygous patients with Fabry's disease. In: Kidney International . Volume 13, Number 3, March 1978, pp. 223-235, PMID 418264 .
- ↑ J. Alroy, S. Sabnis, JB Kopp: Renal pathology in Fabry disease. In: JASN. Volume 13 Suppl 2, June 2002, pp. S134-S138, PMID 12068025 . (Review).
- ↑ a b c d e T. Thomaidis, M. Relle u. a .: Effect of enzyme replacement therapy (ERT) on kidney function in patients with Fabry disease. In: Medical Clinic. Volume 104, Number 9, September 2009, pp. 699-703, doi: 10.1007 / s00063-009-1152-1 . PMID 19779674 . (Review).
- ↑ A. Ortiz, JP Oliveira et al. a .: Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. In: Nephrology, dialysis, transplantation. Volume 23, Number 5, May 2008, pp. 1600-1607, doi: 10.1093 / ndt / gfm848 . PMID 18175781 .
- ↑ a b MH Branton, R. Schiffmann u. a .: Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. In: Medicine. Volume 81, Number 2, March 2002, pp. 122-138, PMID 11889412 .
- ↑ a b c d R. Schiffmann, DG Warnock u. a .: Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. In: Nephrology, dialysis, transplantation. Volume 24, Number 7, July 2009, pp. 2102-2111, doi: 10.1093 / ndt / gfp031 . PMID 19218538 . PMC 2698092 (free full text).
- ^ Glass RB et al. J Comput Assist Tomogr (2004) 28: 158-168
- ↑ a b c A. Linhart, T. Palecek and a .: New insights in cardiac structural changes in patients with Fabry's disease. In: American Heart Journal . Volume 139, number 6, June 2000, pp. 1101-1108, doi: 10.1067 / mhj.2000.105105 . PMID 10827394 .
- ↑ a b c C. Kampmann, F. Baehner u. a .: Cardiac manifestations of Anderson-Fabry disease in heterozygous females. In: Journal of the American College of Cardiology . Volume 40, Number 9, November 2002, pp. 1668-1674, PMID 12427421 .
- ↑ a b M. Senechal, DP Germain: Fabry disease: a functional and anatomical study of cardiac manifestations in 20 hemizygous male patients. In: Clinical genetics. Volume 63, Number 1, January 2003, pp. 46-52, PMID 12519371 .
- ↑ a b JS Shah, DA Hughes u. a .: Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. In: The American journal of cardiology. Volume 96, number 6, September 2005, pp. 842-846, doi: 10.1016 / j.amjcard.2005.05.033 . PMID 16169374 .
- ^ PM Elliott, H. Kindler u. a .: Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. In: Heart. Volume 92, number 3, March 2006, pp. 357-360, doi: 10.1136 / hrt.2004.054015 . PMID 16085718 . PMC 1860797 (free full text).
- ^ JC Moon, M. Sheppard et al. a .: The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. In: J Cardiovasc Magn Reson. Volume 8, Number 3, 2006, pp. 479-482, PMID 16755835 .
- ↑ H. Hasegawa, H. Takano et al. a .: Images in cardiovascular medicine. Transition from left ventricular hypertrophy to massive fibrosis in the cardiac variant of Fabry disease. In: Circulation. Volume 113, number 16, April 2006, pp. E720 – e721, doi: 10.1161 / CIRCULATIONAHA.105.584292 . PMID 16636179 .
- ↑ F. Weidemann, F. Breunig u. a .: The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. In: European heart journal. Volume 26, Number 12, June 2005, pp. 1221-1227, doi: 10.1093 / eurheartj / ehi143 . PMID 15728649 .
- ↑ A. Linhart, C. Kampmann u. a .: Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. In: European heart journal. Volume 28, number 10, May 2007, pp. 1228-1235, doi: 10.1093 / eurheartj / ehm153 . PMID 17483538 .
- ↑ C. Kampmann, A. Linhart u. a .: Onset and progression of the Anderson-Fabry disease related cardiomyopathy. In: International journal of cardiology. Volume 130, number 3, November 2008, pp. 367-373, doi: 10.1016 / j.ijcard.2008.03.007 . PMID 18572264 .
- ↑ a b T. Takenaka, H. Teraguchi and a .: Terminal stage cardiac findings in patients with cardiac Fabry disease: an electrocardiographic, echocardiographic, and autopsy study. In: Journal of cardiology. Volume 51, Number 1, February 2008, pp. 50-59, doi: 10.1016 / j.jjcc.2007.12.001 . PMID 18522775 .
- ↑ a b M. Pieroni, C. Chimenti u. a .: Early detection of Fabry cardiomyopathy by tissue Doppler imaging. In: Circulation. Volume 107, Number 15, April 2003, pp. 1978-1984, doi: 10.1161 / 01.CIR.0000061952.27445.A0 . PMID 12668521 .
- ↑ F. Weidemann, F. Breunig u. a .: Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. In: Circulation. Volume 108, Number 11, September 2003, pp. 1299-1301, doi: 10.1161 / 01.CIR.0000091253.71282.04 . PMID 12952834 .
- ↑ M. Pieroni, C. Chimenti et al. a .: Tissue Doppler imaging in Fabry disease. In: Current opinion in cardiology. Volume 19, Number 5, September 2004, pp. 452-457, PMID 15316452 . (Review).
- ↑ T. Palecek, G. Dostalova et al. a .: Right ventricular involvement in Fabry disease. In: Journal of the American Society of Echocardiography. Volume 21, Number 11, November 2008, pp. 1265-1268, doi: 10.1016 / j.echo.2008.09.002 . PMID 18835697 .
- ↑ M. Niemann, F. Breunig a. a .: The right ventricle in Fabry disease: natural history and impact of enzyme replacement therapy. In: Heart. Volume 96, number 23, December 2010, pp. 1915-1919, doi: 10.1136 / hrt.2010.204586 . PMID 20965976 .
- ↑ C. Kampmann, FA Baehner a. a .: The right ventricle in Fabry disease. In: Acta paediatrica. Volume 94, Number 447, March 2005, pp. 15-18, PMID 15895706 .
- ^ A b A. Fellgiebel, MJ Müller, L. Ginsberg: CNS manifestations of Fabry's disease. In: Lancet neurology. Volume 5, Number 9, September 2006, pp. 791-795, doi: 10.1016 / S1474-4422 (06) 70548-8 . PMID 16914407 .
- ↑ a b c K. Sims, J. Politei u. a .: Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. In: Stroke. Volume 40, number 3, March 2009, pp. 788-794, doi: 10.1161 / STROKEAHA.108.526293 . PMID 19150871 .
- ↑ a b P. Mitsias, SR Levine: Cerebrovascular complications of Fabry's disease. In: Annals of neurology. Volume 40, Number 1, July 1996, pp. 8-17, doi: 10.1002 / ana.410400105 . PMID 8687196 .
- ↑ MF Mendez, TM Stanley et al. a .: The vascular dementia of Fabry's disease. In: Dementia and geriatric cognitive disorders. Volume 8, Number 4, 1997 Jul-Aug, pp. 252-257, PMID 9213072 .
- ↑ R. Okeda, M. Nisihara: An autopsy case of Fabry disease with neuropathological investigation of the pathogenesis of associated dementia. In: Neuropathology. Volume 28, Number 5, October 2008, pp. 532-540, doi: 10.1111 / j.1440-1789.2008.00883.x . PMID 18410273 .
- ↑ A. Fellgiebel, I. Keller a. a .: Diagnostic utility of different MRI and MR angiography measures in Fabry disease. In: Neurology. Volume 72, number 1, January 2009, pp. 63-68, doi: 10.1212 / 01.wnl.0000338566.54190.8a . PMID 19122032 .
- ↑ T. DeGraba, S. Azhar et al. a .: Profile of endothelial and leukocyte activation in Fabry patients. In: Annals of neurology. Volume 47, Number 2, February 2000, pp. 229-233, PMID 10665494 .
- ^ DF Moore, LT Scott et al. a .: Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. In: Circulation . Volume 104, Number 13, September 2001, pp. 1506-1512, PMID 11571244 .
- ↑ DF Moore, G. Altarescu et al. a .: Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. In: Stroke; a journal of cerebral circulation. Volume 33, Number 2, February 2002, pp. 525-531, PMID 11823664 .
- ↑ R. Schiffmann: Fabry disease. In: Pharmacology & Therapeutics . Volume 122, number 1, April 2009, pp. 65-77, doi: 10.1016 / j.pharmthera.2009.01.003 . PMID 19318041 . (Review).
- ↑ CR Kaneski, DF Moore a. a .: Myeloperoxidase predicts risk of vasculopathic events in hemizgygous males with Fabry disease. In: Neurology. Volume 67, number 11, December 2006, pp. 2045-2047, doi: 10.1212 / 01.wnl.0000247278.88077.09 . PMID 17159117 . PMC 1950664 (free full text).
- ↑ A. Palla, S. Hegemann u. a .: Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy. In: Journal of neurology. Volume 254, Number 10, October 2007, pp. 1433-1442, doi: 10.1007 / s00415-007-0575-y . PMID 17934877 .
- ↑ Y. Sakurai, H. Kojima et al. a .: The hearing status in 12 female and 15 male Japanese Fabry patients. In: auris, nasus, larynx. Volume 36, number 6, December 2009, pp. 627-632, doi: 10.1016 / j.anl.2009.01.001 . PMID 19261412 .
- ↑ DP Germain, P. Avan et al. a .: Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. In: BMC medical genetics. Volume 3, October 2002, p. 10, PMID 12377100 . PMC 134464 (free full text).
- ^ G. Conti, B. Sergi: Auditory and vestibular findings in Fabry disease: a study of hemizygous males and heterozygous females. In: Acta paediatrica. Volume 92, Number 443, December 2003, pp. 33-37, PMID 14989464 .
- ↑ DM Rosenberg, VJ Ferrans u. a .: Chronic airflow obstruction in Fabry's disease. In: The American journal of medicine. Volume 68, Number 6, June 1980, pp. 898-905, PMID 6247911 .
- ↑ LK Brown, A. Miller et al. a .: Pulmonary involvement in Fabry disease. In: American journal of respiratory and critical care medicine. Volume 155, Number 3, March 1997, pp. 1004-1010, PMID 9116979 .
- ↑ S. Magage, JC Lubanda et al. a .: Atteinte respiratoire de la maladie de Fabry. In: Médecine sciences. Volume 21, Number 11 Suppl, December 2005, pp. 37-39, doi: 10.1051 / medsci / 20052111s37 . PMID 16324666 .
- ↑ S. Magage, JC Lubanda et al. a .: Natural history of the respiratory involvement in Anderson-Fabry disease. In: Journal of inherited metabolic disease. Volume 30, Number 5, October 2007, pp. 790-799, doi: 10.1007 / s10545-007-0616-9 . PMID 17619837 .
- ↑ DP Germain, K. Benistan u. a .: Atteinte osseuse de la maladie de Fabry. In: Médecine sciences. Volume 21, Number 11 Suppl, December 2005, pp. 43-44, doi: 10.1051 / medsci / 20052111s43 . PMID 16324668 .
- ↑ DP Germain, K. Benistan u. a .: Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. In: Clinical genetics. Volume 68, Number 1, July 2005, pp. 93-95, doi: 10.1111 / j.1399-0004.2005.00457.x . PMID 15952993 .
- ↑ H. Mersebach, JO Johansson u. a .: Osteopenia: a common aspect of Fabry disease. Predictors of bone mineral density. In: Genetics in medicine. Volume 9, Number 12, December 2007, pp. 812-818, doi: 10.1097 / GIM.0b013e31815cb197 . PMID 18091430 .
- ^ RO Brady : Bone and muscle involvement in Fabry disease. In: D. Elstein, G. Altarescu, M. Beck (Eds.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , pp. 293-298, limited preview in the Google book search
- ↑ CL Marchesoni, N. Roa et al. a .: Misdiagnosis in Fabry disease. In: The Journal of pediatrics. Volume 156, Number 5, May 2010, pp. 828-831, doi: 10.1016 / j.jpeds.2010.02.012 . PMID 20385321 .
- ↑ A. Mehta, R. Ricci et al. a .: Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. In: European journal of clinical investigation. Volume 34, Number 3, March 2004, pp. 236-242, doi: 10.1111 / j.1365-2362.2004.01309.x . PMID 15025684 .
- ↑ a b c KD MacDermot, A. Holmes, AH Miners: Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. In: Journal of medical genetics. Volume 38, Number 11, November 2001, pp. 750-760, PMID 11694547 . PMC 173476 (free full text).
- ↑ N. Siegmund-Schultze: Fabry disease is rare, the symptoms are unspecific - an odyssey from doctor to doctor often precedes the diagnosis. In: Doctors newspaper. dated December 15, 2004
- ↑ JS Mayes, JB Scheerer et al. a .: Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry's disease. In: Clinica chimica acta. Volume 112, Number 2, May 1981, pp. 247-251, PMID 6263521 .
- ↑ B. Hoffmann, H. Georg Koch a. a .: Deficient alpha-galactosidase A activity in plasma but no Fabry disease - a pitfall in diagnosis. In: Clinical chemistry and laboratory medicine. Volume 43, number 11, 2005, pp. 1276-1277, doi: 10.1515 / CCLM.2005.219 . PMID 16232095 .
- ↑ GE Linthorst, AC Vedder u. a .: Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. In: Clinica chimica acta. Volume 353, number 1–2, March 2005, pp. 201–203, doi: 10.1016 / j.cccn.2004.10.019 . PMID 15698608 .
- ↑ a b BE Smid, L van der Tol, M Biegstraaten, GE Linthorst, CEM Hollak, BJHM Poorthuis. In: J Med Genet , 2015, 52 (4), pp. 262-226
- ^ S. Roy, K. Gaudin et al. a .: Optimization of the separation of four major neutral glycosphingolipids: application to a rapid and simple detection of urinary globotriaosylceramide in Fabry disease. In: Journal of chromatography B. Volume 805, Number 2, June 2004, pp. 331-337, doi: 10.1016 / j.jchromb.2004.03.037 . PMID 15135109 .
- ↑ C. Auray-Blais, D. Cyr et al. a .: Development of a filter paper method potentially applicable to mass and high-risk urinary screenings for Fabry disease. In: Journal of inherited metabolic disease. Volume 30, Number 1, February 2007, p. 106, doi: 10.1007 / s10545-006-0444-3 . PMID 17171433 .
- ↑ C. Auray-Blais, D. Cyr et al. a .: Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. In: Molecular Genetics and Metabolism. Volume 93, Number 3, March 2008, pp. 331-340, doi: 10.1016 / j.ymgme.2007.10.001 . PMID 18023222 .
- ↑ D. Touboul, S. Roy et al. a .: Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. In: Analytical and bioanalytical chemistry. Volume 382, Number 5, July 2005, pp. 1209-1216, doi: 10.1007 / s00216-005-3239-8 . PMID 15959771 .
- ↑ K. Mills, P. Morris et al. a .: Measurement of urinary CDH and CTH by tandem mass spectrometry in patients hemizygous and heterozygous for Fabry disease. In: Journal of inherited metabolic disease. Volume 28, Number 1, 2005, pp. 35-48, doi: 10.1007 / s10545-005-5263-4 . PMID 15702404 .
- ↑ RJ Desnick: Prenatal diagnosis of Fabry disease. In: Prenatal diagnosis. Volume 27, Number 8, August 2007, pp. 693-694, doi: 10.1002 / pd.1767 . PMID 17533632 . (Review).
- ↑ C. Lindengrün: rare diseases - faces of mucopolysaccharidoses. In: CliniCum. 9, 2010.
- ↑ AM Das: Congenital metabolic disorders. In: A. Rieder, B. Lohff (Eds.): Gender Medicine. Verlag Springer, 2004, ISBN 3-211-00766-0 , p. 67f. limited preview in Google Book search
- ↑ TF Metz, TP Mechtler u. a .: Simplified newborn screening protocol for lysosomal storage disorders. In: Clinical chemistry. Volume 57, number 9, September 2011, pp. 1286-1294, doi: 10.1373 / clinchem.2011.164640 . PMID 21771947 .
- ↑ K. Nakamura, K. Hattori, F. Endo: Newborn screening for lysosomal storage disorders. In: American journal of medical genetics. Volume 157, number 1, February 2011, pp. 63-71, doi: 10.1002 / ajmg.c.30291 . PMID 21312327 . (Review).
- ↑ Spada M, Kasper D, Pagliardini V, Biamino E, Giachero S, Porta F. Ital J Pediatr 2017; 43 (1): 1
- ^ GM Pastores: Miglustat: Substrate Reduction Therapy for Lysosomal Storage Disorders Associated with Primary Central Nervous System Involvement. ( Memento from June 16, 2010 in the Internet Archive ) (PDF; 75 kB) In: Recent Patents on CNS Drug Discovery. 1, 2006, pp. 77-82. PMID 18221193
- ↑ a b Therapy of Fabry's disease. ( Memento of February 3, 2015 in the Internet Archive ) Retrieved September 13, 2011
- ↑ JM Donnelly: Shire wins FDA approval for rare disease drug. In: Boston Business Journal. dated August 25, 2011
- ↑ a b CA Black: March on, not in. In: Nature medicine. Volume 17, number 5, May 2011, p. 515, doi: 10.1038 / nm0511-515 . PMID 21546944 .
- ↑ a b M. Beck: Agalsidase alfa - a preparation for enzyme replacement therapy in Anderson-Fabry disease. In: Expert opinion on investigational drugs. Volume 11, Number 6, June 2002, pp. 851-858, doi: 10.1517 / 13543784.11.6.851 . PMID 12036428 . (Review).
- ↑ K. Lee, X. Jin et al. a .: A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. In: Glycobiology. Volume 13, Number 4, April 2003, pp. 305-313, doi: 10.1093 / glycob / cwg034 . PMID 12626384 .
- ↑ Specialist information Fabrazyme 5mg, 35mg, as of 11/2018
- ↑ a b WR Wilcox, M. Banikazemi u. a .: Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. In: American journal of human genetics. Volume 75, Number 1, July 2004, pp. 65-74, doi: 10.1086 / 422366 . PMID 15154115 . PMC 1182009 (free full text).
- ↑ BL Thurberg, H. Rennke u. a .: Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. In: Kidney International . Volume 62, Number 6, December 2002, pp. 1933-1946, doi: 10.1046 / j.1523-1755.2002.00675.x . PMID 12427118 .
- ↑ a b c d J. Marshall, KM Ashe u. a .: Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. In: PloS one. Volume 5, number 11, 2010, p. E15033, doi: 10.1371 / journal.pone.0015033 . PMID 21124789 . PMC 2991350 (free full text). ( Open access )
- ↑ M. Beck: Agalsidase alfa for the treatment of Fabry disease: new data on clinical efficacy and safety. In: Expert opinion on biological therapy. Volume 9, Number 2, February 2009, pp. 255-261, doi: 10.1517 / 14712590802658428 . PMID 19236256 . (Review).
- ↑ a b CM Eng, N. Guffon et al. a .: Safety and efficacy of recombinant human alpha-galactosidase A - replacement therapy in Fabry's disease. In: The New England Journal of Medicine . Volume 345, Number 1, July 2001, pp. 9-16, doi: 10.1056 / NEJM200107053450102 . PMID 11439963 .
- ↑ GM Keating, D. Simpson: Spotlight on agalsidase beta in Fabry disease. In: BioDrugs. Volume 21, Number 4, 2007, pp. 269-271, PMID 17628124 . (Review).
- ↑ DP Germain, S. Waldek u. a .: Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. In: JASN. Volume 18, Number 5, May 2007, pp. 1547–1557, doi: 10.1681 / ASN.2006080816 . PMID 17409312 .
- ↑ a b Y. Tajima, I. Kawashima et al. a .: Use of a modified alpha-N-acetylgalactosaminidase in the development of enzyme replacement therapy for Fabry disease. In: American journal of human genetics. Volume 85, Number 5, November 2009, pp. 569-580, doi: 10.1016 / j.ajhg.2009.09.016 . PMID 19853240 . PMC 2775840 (free full text).
- ↑ T. Ohashi, M. Sakuma et al. a .: Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. In: Molecular Genetics and Metabolism. Volume 92, Number 3, November 2007, pp. 271-273, doi: 10.1016 / j.ymgme.2007.06.013 . PMID 17689998 .
- ↑ T. Ohashi, S. Iizuka et al. a .: Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. In: Molecular Genetics and Metabolism. Volume 94, number 3, July 2008, pp. 313-318, doi: 10.1016 / j.ymgme.2008.03.008 . PMID 18456533 .
- ↑ Lenders et al., J Allergy and Clin. Immunology, 2018
- ↑ Maarten Arends, Marieke Biegstraaten u. a .: Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. In: Journal of Medical Genetics , 55, 2018, p. 351, doi: 10.1136 / jmedgenet-2017-104863 .
- ^ Pharmaceutical Newspaper, Issue 05 2016
- ↑ a b c SD James: Fabry Disease Patients Get Sicker as Drugs Go Overseas. In: abcNews.com. dated August 30, 2011
- ↑ CA Black: To march in or not to march in. In: Nature medicine. Volume 17, number 8, 2011, p. 921, doi: 10.1038 / nm.2440 . PMID 21818082 .
- ↑ rme: EMEA: Rationing of Cerezyme and Fabrazyme. ( Page no longer available , search in web archives ) Info: The link was automatically marked as defective. Please check the link according to the instructions and then remove this notice. In: Aerzteblatt.de. dated June 28, 2009
- ↑ Supply shortages of Cerezyme and Fabrazyme - priority access for patients most in need of treatment recommended. (PDF; 35 kB) EMEA press release of June 25, 2009
- ↑ G. Schuh, C. Nussbaum u. a .: Dealing with know-how in international R&D cooperation. (PDF; 1.8 MB) In: Federal Ministry for Education and Research (Ed.), 2009, p. 21.
- ^ A b E. Dolgin: NIH faces marching orders on orphan drug shortage. In: Nature medicine. Volume 17, Number 5, May 2011, p. 522, doi: 10.1038 / nm0511-522b . PMID 21546953 .
- ↑ M. Ratner: Fabry drug march-in denied. In: Nature biotechnology . Volume 29, number 97, 2011, p. 676, doi: 10.1038 / nbt0211-97b .
- ^ FS Collins: Determination in the Case of Fabrazyme Manufactured by Genzyme Corporation. (PDF; 544 kB) From December 1, 2010
- ^ T. Staton: Genzyme apologizes for latest Fabrazyme delay. In: FiercePharma. dated September 8, 2011
- ↑ T. Staton: Quality-control issue disrupts Fabrazyme flow. In: FiercePharma. dated August 5, 2011
- ↑ a b Sanofi's Genzyme starts shipping Fabry disease drug. In: Pharma Times. March 2, 2012, accessed April 30, 2019 .
- ↑ a b Shire withdraws application for approval from FDA. Doctors newspaper , March 16, 2012, accessed on April 29, 2019 .
- ↑ Shire withdraws Replagal in USA as FDA wants more trials. Pharma Times, March 15, 2012, accessed April 29, 2019 .
- ↑ Timeline for Fabrazyme, Replagal *. (PDF) July 14, 2014, accessed April 29, 2019 .
- ↑ U. Fricke, U. Schwabe: Neue Arzneimittel 2007. In: U. Schwabe, D. Paffrath (Ed.): Drug Ordinance Report 2008. Verlag Springer, 2008, p. 74. Restricted preview in the Google book search
- ↑ B. Hoffmann, E. Mayatepek: Fabry disease-often seen, rarely diagnosed. In: Deutsches Ärzteblatt international. Volume 106, number 26, June 2009, pp. 440–447, doi: 10.3238 / arztebl.2009.0440 . PMID 19623315 . PMC 270439 (free full text). (Review).
- ↑ T. Watt, AP Burlina a. a .: Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. In: Genetics in medicine. Volume 12, Number 11, November 2010, pp. 703-712, doi: 10.1097 / GIM.0b013e3181f13a4a . PMID 20885332 .
- ↑ MR Filling-Katz, HF Merrick u. a .: Carbamazepine in Fabry's disease: effective analgesia with dose-dependent exacerbation of autonomic dysfunction. In: Neurology . Volume 39, Number 4, April 1989, pp. 598-600, PMID 2494569 .
- ↑ National Institute for Health and Clinical Excellence: Neuropathic pain: The pharmacological management of neuropathic pain in adults in non-specialist settings (NICE clinical guideline 96). Center for Clinical Practice at NICE; 2010.
- ^ R. Baron, A. Binder, G. Wasner: Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. In: Lancet neurology. Volume 9, Number 8, August 2010, pp. 807-819, doi: 10.1016 / S1474-4422 (10) 70143-5 . PMID 20650402 . (Review).
- ↑ G. Cruccu, C. Sommer a. a .: EFNS guidelines on neuropathic pain assessment: revised 2009. In: European journal of neurology. Volume 17, number 8, August 2010, pp. 1010-1018, doi: 10.1111 / j.1468-1331.2010.02969.x . PMID 20298428 . (Review).
- ↑ CE Argoff, NW Barton u. a .: Gastrointestinal symptoms and delayed gastric emptying in Fabry's disease: response to metoclopramide. In: Nuclear medicine communications. Volume 19, Number 9, September 1998, pp. 887-891, PMID 10581595 .
- ↑ H. Tahir, LL Jackson, DG Warnock: Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. In: JASN. Volume 18, number 9, September 2007, pp. 2609-2617, doi: 10.1681 / ASN.2006121400 . PMID 17656478 .
- ↑ a b CE Stetter: In vivo investigation of the cardiac metabolism in Fabry disease using 31 phosphorus MR spectroscopy. Dissertation, Julius Maximilians University of Würzburg, 2007.
- ↑ IB Tomasic, MC Metcalf u. a .: Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases. In: The Journal of biological chemistry . Volume 285, Number 28, July 2010, pp. 21560-21566, doi: 10.1074 / jbc.M110.118588 . PMID 20444686 . PMC 2898384 (free full text).
- ↑ Galafold. In: EMA Europe. September 17, 2018, accessed April 2, 2019 .
- ^ S. Ishii, HH Chang et al. a .: Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. In: The Biochemical journal. Volume 406, Number 2, September 2007, pp. 285-295, doi: 10.1042 / BJ20070479 . PMID 17555407 . PMC 1948963 (free full text).
- ^ S. Ishii, HH Chang et al. a .: Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. In: The Journal of pharmacology and experimental therapeutics. Volume 328, Number 3, March 2009, pp. 723-731, doi: 10.1124 / jpet.108.149054 . PMID 19106170 .
- ↑ R. Hamanaka, T. Shinohara et al. a .: Rescue of mutant alpha-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. In: Biochimica et biophysica acta . Volume 1782, number 6, June 2008, pp. 408-413, doi: 10.1016 / j.bbadis.2008.03.001 . PMID 18381081 .
- ↑ a b S. Biastoff, B. Dräger: Calystegines. In: GA Cordell (Ed.): The Alkaloids: Chemistry and Biology. P. 91. Limited preview in Google Book search
- ↑ GH Yam, N. Bosshard et al. a .: Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. In: American journal of physiology. Volume 290, Number 4, April 2006, pp. C1076-C1082, doi: 10.1152 / ajpcell.00426.2005 . PMID 16531566 .
- ↑ N. Asano, S. Ishii et al. a .: In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. In: European Journal of Biochemistry . Volume 267, Number 13, July 2000, pp. 4179-4186, PMID 10866822 .
- ↑ a b G. Andreotti, MR Guarracino u. a .: Prediction of the responsiveness to pharmacological chaperones: lysosomal human alpha-galactosidase, a case of study. In: Orphanet Journal of Rare Diseases. Volume 5, 2010, p. 36, doi: 10.1186 / 1750-1172-5-36 . PMID 21138548 . PMC 3016270 (free full text). ( Open access )
- ↑ R. Khanna, R. Soska et al. a .: The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. In: Molecular therapy. Volume 18, number 1, January 2010, pp. 23-33, doi: 10.1038 / mt.2009.220 . PMID 19773742 . PMC 2839206 (free full text).
- ↑ Clinical study (phase III): Study of the Effects of Oral AT1001 (Migalastat Hydrochloride) in Patients With Fabry Disease at Clinicaltrials.gov of the NIH
- ↑ Decision of the Commission of 22-V-2006 on the designation of the drug "1-deoxygalactonojirimycin hydrochloride" as an orphan drug according to Regulation (EC) No. 141/2000 of the European Parliament and of the Council. (PDF; 17 kB) (PDF) May 22, 2006
- ↑ SH Shin, S. Kluepfel-Stahl u. a .: Prediction of response of mutated alpha-galactosidase A to a pharmacological chaperone. In: Pharmacogenetics and genomics. Volume 18, Number 9, September 2008, pp. 773-780, doi: 10.1097 / FPC.0b013e32830500f4 . PMID 18698230 . PMC 2657085 (free full text).
- ↑ A. Abe, S. Gregory et al. a .: Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. In: The Journal of clinical investigation. Volume 105, Number 11, June 2000, pp. 1563-1571, doi: 10.1172 / JCI9711 . PMID 10841515 . PMC 300859 (free full text).
- ↑ a b c T. Heare, NJ Alp u. a .: Severe endothelial dysfunction in the aorta of a mouse model of Fabry disease; partial prevention by N-butyldeoxynojirimycin treatment. In: Journal of inherited metabolic disease. Volume 30, Number 1, February 2007, pp. 79-87, doi: 10.1007 / s10545-006-0473-y . PMID 17189993 .
- ↑ EMA Europe: Zavesca. September 17, 2018, accessed April 2, 2019 .
- ↑ GM Pastores, NL Barnett, EH Kolodny: An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. In: Clinical therapeutics. Volume 27, Number 8, August 2005, pp. 1215-1227, doi: 10.1016 / j.clinthera.2005.08.004 . PMID 16199246 .
- ↑ CE Hollak, D. Hughes et al. a .: Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorization safety surveillance program. In: Pharmacoepidemiology and drug safety. Volume 18, Number 9, September 2009, pp. 770-777, doi: 10.1002 / pds.1779 . PMID 19507165 .
- ↑ a b T. Ohshima, R. Schiffmann u. a .: Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. In: PNAS. Volume 96, Number 11, May 1999, pp. 6423-6427, PMID 10339603 . PMC 26897 (free full text).
- ↑ T. Takenaka, GJ Murray et al. a .: Long-term enzyme correction and lipid reduction in multiple organs of primary and secondary transplanted Fabry mice receiving transduced bone marrow cells. In: PNAS. Volume 97, Number 13, June 2000, pp. 7515-7520, doi: 10.1073 / pnas.120177997 . PMID 10840053 . PMC 16577 (free full text).
- ↑ T. Yokoi, H. Kobayashi et al. a .: Minimum requirement of donor cells to reduce the glycolipid storage following bone marrow transplantation in a murine model of Fabry disease. In: The journal of gene medicine. Volume 13, Number 5, May 2011, pp. 262-268, doi: 10.1002 / jgm.1566 . PMID 21520359 .
- ↑ SH Kim, PD Robbins: Retroviral vectors for gene therapy. In: JA Barranger, MA Cabrera-Salazar (Ed.): Lysosomal storage disorders. Verlag Springer, 2007, ISBN 978-0-387-70908-6 , pp. 53-63. limited preview in Google Book search
- ↑ J. Park, GJ Murray et al. a .: Long-term correction of globotriaosylceramide storage in Fabry mice by recombinant adeno-associated virus-mediated gene transfer. In: PNAS. Volume 100, Number 6, March 2003, pp. 3450-3454, doi: 10.1073 / pnas.0537900100 . PMID 12624185 . PMC 152313 (free full text).
- ↑ G. Qin, T. Takenaka et al. a .: Preselective gene therapy for Fabry disease. In: PNAS. Volume 98, Number 6, March 2001, pp. 3428-3433, doi: 10.1073 / pnas.061020598 . PMID 11248095 . PMC 30670 (free full text).
- ^ JE Scherberich: Hereditary kidney diseases. In: G. Steinbeck, G. Paumgartner: Therapy of internal diseases. Verlag Springer, 2005, ISBN 3-540-23750-X , p. 576. Restricted preview in the Google book search
- ↑ YA Ioannou, KM Zeidner u. a .: Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. In: American journal of human genetics. Volume 68, Number 1, January 2001, pp. 14-25, doi: 10.1086 / 316953 . PMID 11115376 . PMC 1234907 (free full text).
- ^ ME Haskins, U. Giger, DF Patterson: Animal models of lysosomal storage diseases: their development and clinical relevance. In: A. Mehta, M. Beck, G. Sunder-Plassmann (Eds.): Fabry Disease: Perspectives from 5 Years of FOS. Chapter 6, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290677
- ↑ a b KD MacDermot, A. Holmes, AH Miners: Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. In: Journal of medical genetics. Volume 38, Number 11, November 2001, pp. 769-775, PMID 11732485 . PMC 1734754 (free full text).
- ↑ A. Mehta, JT Clarke et al. a .: Natural course of Fabry disease: changing pattern of causes of death in FOS - Fabry Outcome Survey. In: Journal of medical genetics. Volume 46, Number 8, August 2009, pp. 548-552, doi: 10.1136 / jmg.2008.065904 . PMID 19473999 .
- ^ A b c d e H. Fabry: An historical overview of Fabry disease. In: Journal of inherited metabolic disease. Volume 24 Suppl 2, 2001, pp. 3-7, PMID 11758677 .
- ↑ a b c J. Fabry: A contribution to the knowledge of the purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). In: Archives for Dermatology and Syphilis . Volume 42, 1898, pp. 187-200. doi : 10.1007 / BF01986897
- ↑ a b c T. Jansen, FG Bechara, P. Altmeyer: Angiokeratome - Clinic, Diagnostics and Therapy. TKT - Europe 5S, 2003, ISBN 3-00-011357-6 .
- ↑ a b J. Fabry: On the clinic and etiology of the angiokeratoma. In: Archives for Dermatology and Syphilis. Volume 123, Number 2, 1916, pp. 294-307.
- ↑ J. Fabry: Another contribution to the clinic of Angiokeratoma naeviforme (Naevus angiokeratosus). In: Dermacol Wochenschr. 90, 1930, pp. 339-341.
- ↑ W. Anderson: A case of "Angeio-keratoma". In: Br J Dermatol. Volume 10, 1898, pp. 113-117. doi: 10.1111 / j.1365-2133.1898.tb16317.x
- ^ HJ Wallace: Angiokeratoma corporis diffusum. In: Br J Dermatol. 70, 1968, pp. 354-360. doi: 10.1111 / j.1365-2133.1958.tb13796.x PMID 13584689
- ↑ W. Cottle: Warty growths. In: St. Georges Hospital Reports. 9, 1877-1878, pp. 753-762.
- ↑ J. Weicksel: Angiomatosis or Angiokeratosis universalis (a very rare skin and vascular disease). In: Dtsch Med Wochenschr. Volume 51, 1925, pp. 898-900.
- ↑ AW Pumps, M. Ruiter, HJ Wyers: , as a sign of disease Angiokeratoma corporis diffusum (universal) Fabry of unknown internal; two autopsy reports. In: Acta medica Scandinavica. Volume 128, Number 3, June 1947, pp. 234-255, PMID 18897399 .
- ^ P. Reuter: Springer large dictionary medicine. Verlag Springer, 2004, ISBN 3-540-21352-X , p. 1186. Limited preview in the Google book search
- ↑ K. Scriba: On the pathogenesis of Angiokeratoma corporis diffusum Fabry with cardio-vasorenal symptom complex. In: Verh Dtsch Ges Pathol. 34, 1950, pp. 221-226.
- ↑ H. Hornbostel, K. Scriba: For the diagnosis of angiokeratoma Fabry with cardiovasorenal symptom complex as a phosphatide storage disease by excision of the skin. In: Clinical weekly. Volume 31, Numbers 3-4, January 1953, pp. 68-69, PMID 13062573 .
- ↑ Colley JR, DL Miller et al. a .: The renal lesion in angiokeratoma corporis diffusum. In: British medical journal. Volume 1, Number 5082, May 1958, pp. 1266-1268, PMID 13536474 . PMC 2028866 (free full text).
- ↑ CC Sweeley, B. Klionsky: Fabry's disease. Classification as sphingolipidosis and partial characterization of a novel glycolipid. In: The Journal of biological chemistry. Volume 238, September 1963, pp. 3148-3150, PMID 14081947 .
- ↑ JM Opitz, FC Stiles u. a .: The Genetics of Angiokeratoma Corporis Diffusum (Fabry's Disease) and Its Linkage Relations with the Xg Locus. In: American journal of human genetics. Volume 17, Number 4, July 1965, pp. 325-342, PMID 17948499 . PMC 1932618 (free full text).
- ^ K. Hashimoto, BG Gross, WF Lever: Angiokeratoma Corporis Diffusum (Fabry). Histochemical and Electron Microscopic Studies of the Skin. In: The Journal of investigative dermatology. Volume 44, February 1965, pp. 119-128, doi: 10.1038 / jid.1965.22 PMID 14258908 .
- ↑ M. Niemann: The influence of long-term enzyme replacement therapy on the morphology and function of the left ventricle in patients with Fabry disease. Dissertation, Julius Maximilians University of Würzburg, 2009.
- ^ RO Brady , AE Gal et al. a .: Enzymatic defect in Fabry's disease. Ceramide trihexosidase deficiency. In: The New England journal of medicine . Volume 276, Number 21, May 1967, pp. 1163-1167, doi: 10.1056 / NEJM196705252762101 . PMID 6023233 .
- ↑ JA Kint: Fabry's disease: alpha-galactosidase deficiency. In: Science . Volume 167, Number 922, February 1970, pp. 1268-1269, PMID 5411915 .
- ↑ JW Kusiak, JM Quirk, RO Brady : Purification and properties of the two major isozymes of alpha-galactosidase from human placenta. In: The Journal of biological chemistry . Volume 253, Number 1, January 1978, pp. 184-190, PMID 201618 .
- ↑ RJ Desnick, KY Allen et al. a .: Fabry's disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. In: The Journal of laboratory and clinical medicine. Volume 81, Number 2, February 1973, pp. 157-171, PMID 4683418 .
- ^ RO Brady , JF Tallman et al. a .: Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry's disease. In: The New England journal of medicine . Volume 289, Number 1, July 1973, pp. 9-14, doi: 10.1056 / NEJM197307052890103 . PMID 4196713 .
- ↑ RJ Desnick, KJ Dean et al. a .: Enzyme therapy in Fabry disease: differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. In: PNAS . Volume 76, Number 10, October 1979, pp. 5326-5330, PMID 228284 . PMC 413135 (free full text).
- ↑ CA Mapes, RL Anderson et al. a .: Enzyme replacement in Fabry's disease, an inborn error of metabolism. In: Science . Volume 169, Number 949, September 1970, pp. 987-989, PMID 4914726 .
- ↑ T. Ohshima, GJ Murray et al. a .: alpha-galactosidase A deficient mice: a model of Fabry disease. In: PNAS. Volume 94, Number 6, March 1997, pp. 2540-2544, PMID 9122231 . PMC 20124 (free full text).
- ↑ DF Bishop, DH Calhoun a. a .: Human alpha-galactosidase A: nucleotide sequence of a cDNA clone encoding the mature enzyme. In: PNAS. Volume 83, Number 13, July 1986, pp. 4859-4863, PMID 3014515 . PMC 323842 (free full text).
- ↑ R. Kornreich, RJ Desnick, DF Bishop: Nucleotide sequence of the human alpha-galactosidase A gene. In: Nucleic acids research . Volume 17, Number 8, April 1989, pp. 3301-3302, PMID 2542896 . PMC 317741 (free full text).
- ↑ DF Bishop, R. Kornreich, RJ Desnick: Structural organization of the human alpha-galactosidase A gene: further evidence for the absence of a 3 'untranslated region. In: PNAS. Volume 85, Number 11, June 1988, pp. 3903-3907, PMID 2836863 . PMC 280328 (free full text).
- ↑ R. Schiffmann, GJ Murray u. a .: Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. In: PNAS. Volume 97, Number 1, January 2000, pp. 365-370, PMID 10618424 . PMC 26669 (free full text).
- ↑ Agalsidase beta (Fabrazyme® dry substance; Genzyme GmbH). Retrieved September 1, 2011
- ↑ Agalsidase alfa (Replagal® infusion solution; TKT Europe). Retrieved September 1, 2011