
Atomic absorption spectrometry


The atomic absorption spectrometry ( AAS ) is a to the group of atomic spectrometry belonging analytical method. In analytical chemistry it is a proven and fast method for the quantitative and qualitative analysis of many elements ( metals , semi-metals ) in mostly aqueous solutions and solids. The AAS is based on the attenuation ( absorption ) of radiation through interaction with free atoms. Since every chemical element has a characteristic line spectrum , statements about the elements contained in a sample can be made by evaluating the difference spectrum to a reference measurement without a sample. Atomic absorption spectrometry is divided into the following sub-processes with regard to the conversion of a sample into the gas phase:
- F-AAS (short for flame atomic absorption spectrometry , also known as flame atomic absorption spectrometry )
- GF-AAS or etA-AAS (Graphite furnace atomic absorption spectrometry , German: AAS with electrothermal heating, also called graphite furnace technology )
- CV-AAS ( cold vapor atomic absorption spectrometry , dt. 'AAS mit Kaltdampftechnik', also called hydride technology)
- HR-CS-AAS (engl. High-resolution continuum-source atomic absorption spectrometry , dt. 'Atomic absorption spectrometry with continuum radiator and high-resolution echelle double monochromator')
It was developed by Alan Walsh in Australia in the 1950s.
principle

A light source emits light of different wavelengths with a certain intensity. In the beam path there is an atomization unit in which the components of a sample to be examined are atomized, i.e. i.e. converted into individual, excitable atoms. The elements are atomized either by a gas flame ( ethyne / air or ethyne / nitrous oxide mixture), into which the solution to be analyzed is atomized, or by rapid, strong heating in an electrically heated graphite tube , into which a small amount of the solution is previously placed was added.
After the light beam has been weakened in the atomic cloud ( absorption ), its intensity is measured behind the atomization unit and compared with the intensity of the non-weakened light. It is detected how much of the incident light of a certain wavelength was absorbed by the element to be measured (in most cases the AAS is a single-element technology). The Lambert-Beer law applies . As the concentration of the analyte in the sample increases, the attenuation of the incident light ( extinction ) increases proportionally.
The absorbed light energy is emitted again by the excited atom at the same wavelength , the atom thus shows fluorescence. The fact that you can measure an intensity weakening, the absorption signal, has a geometric reason: The incident light is focused on a very small solid angle by the optics of the device. The re-emission takes place as a spherical wave over the entire solid angle of 4π. Only a negligibly small proportion of this gets through the exit gap with the light from the lamp.
The AAS is a relative measurement method. According to the Lambert-Beer law (valid for low concentrations), the extinction of calibration standards of known concentrations is recorded, a calibration curve is created and samples with an unknown concentration are recorded against this calibration and the concentration is read off (nowadays evaluated by software). A major advantage of AAS over other spectroscopic methods is the selectivity of the process. The lamps used as light sources emit an element-specific electromagnetic spectrum due to the composition of their illuminant (hollow cathode material, salt in an electrodeless discharge lamp (EDL)), which is specifically absorbed by the same element to be examined. Spectral disturbances are very rare in the AAS. The latest developments available on the market, such as high-resolution continuous source AA spectrometers, on the other hand, work with only one light source. A xenon short-arc lamp as a continuous radiation source covers all elements and all available wavelengths. This radiation source opens up the entire wavelength range relevant for AAS in just one step. The sequential multi-element routine is thus possible, provided that the elements to be determined are to be determined from the same dilution. A novelty are molecular bands that can be used for the evaluation, with which additional elements, such as e.g. B. sulfur or phosphorus can be analyzed. An HR-CS-AAS therefore measures independently of hollow cathode lamps. This offers advantages such as B. shorter preparation times, no lengthy burn-in time of the light source, since drift phenomena are corrected simultaneously. The time gained is put into perspective by the fact that, due to the more complex optics, a significantly longer initialization time of the device is required, which again destroys the real advantage of the AAS, a quick readiness to measure for fast analysis of fewer samples.
construction
A line source (e.g. a hollow cathode lamp , HKL) serves as the light source . A nebulizer is used to create fine droplets of the analyte for effective atomization in the heat of a gas flame . A dispersion unit ( monochromator ) is connected downstream to protect the detector . The detector is usually a photomultiplier .

Light sources
Element-specific lamps are used in the AAS. One distinguishes between

- Hollow cathode lamps with a cathode consisting of the element of the analyte. Alternatively you can also
- Super lamps (additional cathode) or
- Electrode-free discharge lamps (EDL, principle gas discharge lamp ) can be used.
Both of the last-mentioned lamp types offer a higher light intensity, which in particular for elements that absorb in the UV range (arsenic, cadmium, lead, antimony, selenium, bismuth, tellurium, mercury), a better detection limit due to a better signal-to-noise ratio (engl. signal to noise ratio , SNR). Normal hollow cathode lamps show a significant deterioration in the intensity of their emission lines below about 300 nm. Both lamp types require a separate power supply, but modern devices such. T. is already built in.
In the HR-CS-AAS only one radiation source is used, a specially developed xenon short-arc lamp ( xenon gas discharge lamp ) as a continuous radiation source for all elements and all wavelengths over the entire spectral range from 190-900 nm. The lamp has a different electrode shape and works under high pressure. Under these conditions, a hot focal point forms, which reaches a temperature of around 10,000 K. The emission intensity of this lamp is over the entire spectral range by at least a factor of 10, in the far UV by more than a factor of 100 more intense than that of conventional hollow cathode lamps. The radiation intensity in the AAS has no influence on the sensitivity, but it does on the signal / noise ratio.
atomization
The goal of atomic absorption spectroscopy is to convert as high a proportion of atoms as possible into the gaseous state and to generate as few excited or ionized atoms as possible. To do this, the sample has to be vaporized (free of solvents and volatile components) and incinerated and dissociated into free atoms. Flames and graphite furnaces are mainly used for atomization in the AAS. A distinction must be made between F-AAS and GF-AAS. With the F-AAS, the sample is fed in continuously at a constant speed, from which signals that are constant over time are obtained. With the GF-AAS, a known sample quantity is applied only once. In the ideal case, the spectral signal has a maximum and then drops to zero when the atomic cloud is carried out of the atomizer. The same atomizers are used in the HR-CS-AAS as in the classic line source AAS. Due to the visibility of the spectral environment of the analysis line, the method development and optimization can be made easier and easier for an experienced analyst in the HR-CS-AAS, but an inexperienced user can also be unsettled by the additional information.
Flame atomic absorption spectroscopy
In flame atomic absorption spectroscopy (F-AAS), also known as flame technique, the dissolved sample is first converted into an aerosol . For this purpose, the sample is atomized into a mixing chamber with a pneumatic atomizer and swirled with fuel gas and oxidant ( oxidizing agent ). A fine mist, an aerosol, forms. In order to make the droplet size even smaller and more uniform, the aerosol first hits a ceramic ball and then, if necessary, a mixer blade that only allows fine droplets to pass. A small part of the original aerosol eventually ends up in the flame from the mixing chamber. There, the solvent first evaporates and the solid sample components melt, evaporate and finally dissociate. Excessively high flame temperatures can lead to ionization interference, especially with alkali and some alkaline earth elements , which can be controlled by adding an ionization buffer ( cesium or potassium chloride ). Too low a flame temperature leads to chemical interference . In the flame AAS, the flame can alternatively be operated with two different gas mixtures.
- Air-acetylene flame: This flame is usually used, it uses air as the oxidant and acetylene as the fuel gas.
- Laughing gas acetylene flame: The compounds of some elements (e.g. aluminum , silicon , titanium , but also calcium and chromium ) require higher temperatures for dissociation. In this case, the gas nitrous oxide ( laughing gas ) is used as an oxidant instead of compressed air . At approx. 2800 ° C, this flame is around 500 ° C hotter than the air-acetylene flame. Due to their reducing effect, oxides of z. B. chromium, calcium and aluminum are atomized.

The flame photometer FP8800 can determine the concentration of 4 alkali and alkaline earth elements at the same time.

"Normal" nitrous oxide flame

Laughing gas flame with high salt load



Graphite furnace atomic absorption spectrometry

In graphite furnace atomic absorption spectrometry (GF-AAS), also called graphite furnace technology or atomic absorption spectrometry with electrothermal heating (EtA-AAS), one makes use of the fact that graphite conducts the current and heats up due to its electrical resistance when an electrical voltage is applied.
First, 5 to 50 microliters of the sample solution are placed in a graphite furnace and heated in several steps. The program depends largely on the element to be analyzed and its chemical environment. It also plays a major role in what kind of device and in what kind of graphite furnace system (longitudinally heated / cross-heated graphite furnace) is used. In general, it can be said that pyrolysis temperatures that are approx. 200 ° C lower and atomization temperatures 200 ° C to 400 ° C lower are used in cross-heated graphite furnace. The “Recommended Conditions” of the graphite furnace manufacturer should serve as a guide for choosing the correct temperature / time program. Starting from this, temperatures and times should be optimized in such a way that the measurement signal has a maximum signal area with a minimum background signal. The sample composition may make it necessary to deviate from the standard program.
- Drying 1 : The oven is heated to 90 ° C to 130 ° C for about 30 s in order to concentrate the sample and almost dry it.
- Drying 2 : the oven is heated to 400 ° C for about 20 s to completely dry the sample (if crystal water is present)
- Pyrolysis : the oven is heated to 400 ° C to 1500 ° C (depending on the element) for about 30 s to remove the organic components. This is done through pyrolysis or incineration
- Atomization : at 1500 ° C to 2500 ° C (depending on the element-specific atomization temperature) the sample is atomized for about 5 s
- Bakeout : finally, after the end of the analysis, the temperature is raised to between 2500 ° C (cross-heated oven) and 2800 ° C (longitudinally heated oven) for about 3 s in order to atomize any remaining sample
Each step includes a rise time (ramp) within which the specified temperature is reached. The slower the heating rate selected, the lower the risk of sample splashing and the better the precision with several repeat measurements. For gentler drying, the step "Drying 1" can be split up, e.g. B. in a step at 110 ° C and a step at 130 ° C. Step “Drying 2” can also be omitted for simple samples (drinking water); it is more likely to be used for samples with a complex matrix (body fluids or highly salty wastewater). A ramp of 0 seconds is usually chosen for the atomization step, in this case the maximum power of the voltage supply is given to the graphite tube in order to achieve a maximum heating rate. As a result, the atomic cloud of the analyte reaches a maximum density and maximum sensitivity results. The temperatures are of course dependent on the analyte and can vary widely. The advantage over the flame technique is that the sample can be brought quantitatively into the beam path and remains there longer (up to 7 s). Furthermore, interfering matrix components can often be separated by different evaporation temperatures; they either evaporate beforehand or they stay behind. The detection limits are therefore up to three powers of ten better than with flame technology or ICP-OES . However, interference can occur if work is not carried out under specific measurement conditions. The summary of all measures that lead to a trouble-free analysis in the graphite tube AAS is called the STPF concept ( stabilized temperature platform furnace ).
STPF concept
- Pyrolytically coated graphite tube (better durability and sensitivity)
- Platform in the graphite tube (atomization into a temperature-constant gas phase in the graphite tube)
- Peak area evaluation (less dependence on the time of maximum atomization of the analyte)
- Use of modifiers (stabilization of the analyte at higher pyrolysis temperatures or reduction of the decomposition temperature of matrix components)
- Gas stop during atomization (atomic cloud remains in the pipe longer)
- fast signal acquisition
- Cross-heated graphite furnace (constant temperature over the entire length of the tube, no condensation effects and recombination to form molecules)
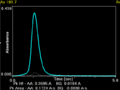
transient (time-resolved) signal from a measurement of arsenic in a graphite furnace

glowing platform in the longitudinally heated graphite furnace

cross-heated Zeeman graphite furnace



Hydride and cold vapor techniques
Definition of terms
- HG-AAS: hydride generation -AAS hydride technology
- CV-AAS: Cold-Vapor -AAS-Kaltdampftechnik
- HG-ET-AAS: Hydride-Generation-Electrothermal -AAS-Enrichment of hydrides in graphite furnace (also: FIAS-Furnace, FIFU)

- Hydride technology
- For some elements, primarily tin , arsenic , antimony , bismuth , selenium , tellurium and germanium , the relatively simple hydride technique can be used to achieve detection limits comparable to those in a graphite furnace. If the element to be determined forms gaseous hydrides such as AsH 3 , SnH 4 or H 2 Se with nascent hydrogen , these can be carried out of their solution by inert gas (usually argon ) and transferred to a heated glass cuvette. The cuvette is made of quartz glass, since simple glass deforms over time at the temperature used (up to 1000 ° C). The heater can either be an electric heater or the flame of a flame AAS. One advantage of electric heating is the better temperature regulation, since not every element has its maximum sensitivity at the same temperature. In the cell, the hydrides break down again at temperatures around 1200 K into hydrogen and the element to be determined. This reaction is not only temperature-controlled, it also depends on the surface properties of the cell. The hydride technology is not limited to AAS, it is also used in ICP-OES .
- Cold steam technology
- As a form of hydride the cold vapor (CV-AAS) mostly as is to be understood, flow injection mercury vapor compression system (Engl. Flow injection mercury system (FIMS)) executed. Here, with the help of a reducing agent, atomic mercury rather than hydride is generated. The reducing agent can be sodium borohydride (NaBH 4 ) , as above, but tin (II) chloride is more commonly used, which offers greater sensitivity and is less prone to foaming. In the case of mercury, one speaks of cold vapor technology, since the quartz cell does not have to be heated and no activation energy is required for the decomposition of the hydride. Nevertheless, a slight warming to 50 ° C to 100 ° C is advantageous so that no water vapor settles in the cuvette, which can affect the sensitivity.
- FIAS technology
- When hydride or mercury is generated by a flow injection system, many samples can be analyzed in quick succession with little carryover between samples. A defined sample aliquot is pumped into a reactor via a valve, treated with reducing solution and mixed with argon. Before and after the sample aliquot, the system is flushed with acid by switching the valve. The gas produced (hydride or mercury vapor) escapes in a gas-liquid separator, the remaining liquid is pumped out through a waste hose. The gas is transferred to the quartz cell and measured with the spectrometer.
- FIAS furnace technology
- The decomposition of the hydrides can not only take place in the quartz cell, but also in the graphite tube ( Hydride Generation Electrothermal AAS , HG-ET-AAS). For this purpose, the tube has to be covered once with a layer of iridium (solution of IrCl 3 ), on the surface of which the hydride decomposes and the analyte (including mercury) accumulates. The analyte is atomized via a short temperature program without destroying the coating. With the same sample volume, the detection strength is in the range of conventional hydride technology, but can be increased as required with larger volumes. The advantage is the lower susceptibility to failure during the decomposition of the hydrides, which in the quartz cell depends heavily on the surface properties of the cell (impact reaction) and less on the temperature (as long as the activation energy is sufficient). The technique is also known as FIAS furnace technique, FIFU, in combination with a flow injection system.
Monochromator
The monochromator divides the light from the lamp and the luminous atomization unit into its spectrum and isolates a specific wavelength from it. In modern devices, only holographic grids are used, which diffract the incident light . The higher the number of grooves on the grating used, the better the resolution of the optics of a spectrometer. In contrast to emission technologies, AAS uses line sources as radiation sources that do not generate a continuous spectrum, but only the spectral lines of the element contained therein. For this reason, the demands on the resolution of the optics of an AAS are lower than on the optics of an ICP-OES .
The choice of the width of the exit slit is used to limit the range of wavelengths that reach the detector. The use of a continuum radiator in high-resolution continuum source AA spectrometers inevitably requires the use of a high-resolution monochromator. Classic monochromators of this type, as used in optical emission, require a large amount of space and have a strong tendency to wavelength drift. Neither is acceptable in the HR-CS-AAS. The problem was solved with the construction of a compact double monochromator with active wavelength stabilization. Both monochromators are in a Littrow setup with a focal length of 30 and 40 cm. The radiation from the continuum radiator enters the monochromator through the entrance slit and is deflected onto the prism by the first parabolic mirror. The prism is mirrored on the back so that the radiation passes the prism twice before it falls back onto the parabolic mirror, now spectrally broken down. This guides the radiation to the intermediate gap via a deflection mirror. The prism is rotated so that the radiation in the area of the analysis line passes through the gap into the second monochromator. The second parabolic mirror directs the radiation onto the Echelle grating , where the selected spectral range is now highly resolved. The entire high-resolution section of the spectrum is then imaged by the parabolic mirror on the detector. The resolution of the double monochromator is traditionally at a value that is around a factor of 100 better than the resolution of classic AAS devices.

Basic arrangements of monochromators:
- Czerny-Turner grating monochromator : light (A) is focused on the entrance slit (B) and is collimated (parallelized) with a concave mirror (C). The collimated beam is diffracted by a rotatable grating (D) and the dispersed beam is again focused on the exit slit (F) by a second mirror (E). Each wavelength of light is focused on a different place in the gap. The wavelength that is let through the gap (G) depends on the angle of rotation of the grating (D).
- Echelle arrangement: Echelle monochromators combine in atomic spectrometry the advantages of a very compact design with high light throughput and very good optical resolution. The combination of an Echelle grating (splitting the light into its diffraction orders) with a prism (splitting the diffraction orders into the wavelengths) results in a two-dimensional arrangement of the spectrum, which, with a suitable choice of the detector (semiconductor detector with segmentation of the light-sensitive areas), enables simultaneous detection of several Wavelengths. This arrangement is implemented in many OES and in a few AAS.
- Littrow arrangement
detector
Secondary electron multipliers (SEV) or - nowadays increasingly - semiconductor detectors are used to measure the light attenuation . The latter show a more homogeneous and more effective light yield (quantum efficiency) over the wavelength range of interest (190–900 nm) and thus a better signal / noise ratio, which is reflected in better detection limits. A CCD line is used as a detector in the HR-CS-AAS. Each pixel is evaluated independently so that the device works in principle with independent detectors. All pixels are exposed and read out simultaneously. The next exposure takes place during the signal processing, which enables a very quick measurement sequence. The absorption line is essentially covered by five central pixels, while the remaining pixels only show the statistical fluctuations of the baseline. These can be used for correction purposes. After all pixels are exposed and read out simultaneously, all intensity fluctuations that are not dependent on the wavelength, such as fluctuations in the lamp emission, can be determined and eliminated with the aid of correction pixels. This creates an extremely stable and low-noise system that leads to a significant improvement in the signal-to-noise ratio. The same correction system also automatically eliminates any continuous background absorption. The detector not only registers the radiation on the analysis line, but its entire environment. This z. B. spectral interference recognizable and can be avoided more easily.
Interference in the AAS
The presence of accompanying substances in the sample can lead to interference . One differentiates:
- spectral interference
- not spectral interference
Spectral interferences are corrected or at least reduced to a certain degree by background correction. For this purpose, a deuterium lamp (D 2 lamp) is switched into the beam path in the AAS in addition to the radiation source or, alternatively, the Zeeman background correction is used in the graphite furnace AAS . The High-Resolution Continuum Source AAS (HR-CS-AAS) works with a high-resolution Echelle spectrometer. In addition to the intensity of the analysis line, the spectral environment is also registered simultaneously. This means that interference is immediately visible. The need to optimize or correct the parameters is automatically recognized in the HR-CS-AA spectrometers. The use of a high-performance detector also minimizes interference with optimal line separation.
The causes of spectral interference in the AAS are:
- Emission lines of the flame or black body radiation of the glowing graphite furnace (constant light),
- unwanted absorption on the same wavelength
- Scattering on solid smoke particles and gases that are difficult to vaporize ( Rayleigh scattering )
Non-spectral interference ( chemical interference ) occurs during the atomization process. One differentiates:
- Transport interferences are chemical disturbances by matrix components or by physical disturbances by the viscosity, density or the surface tension of the solvent. They are particularly problematic in flame AAS, since only a very small proportion of the sample gets into the flame. The elimination of transport interference is achieved by the standard addition method or a matrix adaptation of the calibration standards to the sample.
- Gas interference occurs when there is no complete dissociation (AB → A + B) or ionization. Complete dissociation can be achieved by adding release agents, for example LaCl 3 for phosphates. Better ionization can be achieved by adding alkali elements. In both cases the mode of action obeys the law of mass action . An excess of this aid causes a shift in the reaction equilibrium to the gaseous analyte in the atomic electronic ground state.
- Evaporation interference plays a role in the graphite tube. They arise from too early or too late evaporation of the analyte in the sample, based on the behavior of the analyte in the calibration standard. As a result, the signal of a sample and a calibration standard can result in a significantly different signal shape and height with the same analyte content. A time-resolved signal (evaluation in the signal area) can still lead to correct results here without having to resort to a standard addition .
Deuterium background correction
When using a deuterium lamp to correct the background, the light attenuation of the hollow cathode lamp and that of a D 2 lamp are recorded either simultaneously or alternately. In contrast to the hollow cathode lamp, the D 2 lamp delivers a continuous light spectrum, the intensity of which depends on the wavelength. Above approx. 350 nm it provides almost no intensity, so that elements with an absorption line above this wavelength can be measured without background correction. The selection of the wavelength range for determining the background absorption is made via the width of the monochromator slit on the spectrometer. As a first approximation, the light from the deuterium lamp is almost only absorbed by the background. The proportion of the attenuation of the analysis wavelength is negligibly small compared to the attenuation of the other wavelengths allowed through by the gap.
During the evaluation, the absorption of the radiation from the D 2 lamp (approximately only background absorption ) is subtracted from the measured radiation from the hollow cathode lamp (total absorption from the background + atomic absorption) . The absorption of the analyte in the sample is obtained. A fundamental error lies in the measurement of the background within a wavelength range, given by the setting of the slit in the monochromator, and not exactly on the analysis wavelength. If the background absorbs particularly strongly next to the actual analysis line, too large an amount of background absorption is deducted from the total absorption. There is a so-called "overcorrection" of the measurement signal with negative measurement results.
Zeeman underground correction
The magnetic field of the Zeeman magnet can be seen as a second "radiation source". When the magnetic field is switched off, the entire light attenuation of the analyte and the substrate is recorded. When the magnetic field is switched on, the Zeeman splitting of the absorption line takes place, so that now the analyte no longer absorbs at the wavelength emitted by the lamp, but only the matrix (the background). The strength of the applied magnetic field is not sufficient for a Zeeman splitting of molecules or particles (of the subsurface). The advantage of this background correction lies in the measurement of the background exactly on the analysis line, which means that the background signal is smaller and can still be measured with little interference even with higher salt loads. The disadvantage is a reduced linearity range and, depending on the element, a reduced sensitivity due to e.g. T. incomplete Zeeman splitting .
Background correction in the HR-CS-AAS
No additional system for background correction is required in the HR-CS-AAS. These devices are equipped with a CCD line and thus, in principle, detectors that work simultaneously and independently. The software selects some of these detectors on both sides of the analysis line and uses them for correction purposes. Any change in radiation intensity that occurs equally on all correction pixels is automatically corrected. These include B. fluctuations in lamp emission, but also any continuous background absorption. Discontinuous underground absorption, e.g. B. direct line superposition with a matrix element or molecular absorption with fine structure can be eliminated mathematically with the help of reference spectra. Broadband and spectral background effects can be separated. The former are automatically corrected using reference pixels and the latter are made visible and thus assessable. In most cases of spectral interference, the excellent resolution is sufficient so that the analysis line can be used for evaluation without being disturbed. With this technique, the workflow is extremely simplified, especially with unknown and changing samples. But the measuring routine is also made easier for routine measurements with a known matrix, since spectral disturbances no longer have to be laboriously corrected. Fully automatic underground routines use the available reference pixels and enable simultaneous correction in real time . The background correction in the HR-CS-AAS offers a large dynamic linear working range, extended detection limits, clear measurement results, eliminates artifacts and corrects in the event of direct line overlap.
Matrix modifiers in the GF-AAS
The use of matrix modifiers that convert the analyte into a uniform, thermally more stable chemical compound (isoforming aid) can also be helpful in this context. As a result, higher temperatures are possible during pyrolysis in order to remove matrix before atomization without prematurely losing the analyte. A mixed modifier made from magnesium nitrate (Mg (NO 3 ) 2 ) and ammonium dihydrogen phosphate (NH 4 H 2 PO 4 ) is often used to determine lead and cadmium . To determine many other elements, the use of a mixed modifier made from palladium (II) nitrate (Pd (NO 3 ) 2 ) and magnesium nitrate, which allows pyrolysis temperatures of around 1000 ° C, is the temperature at which sodium chloride is used in cross-heated graphite tube systems (NaCl), a common component of body fluids, sewage and products of the chemical industry.
advantages and disadvantages
A major advantage of this analytical method is the measurement accuracy, which means that traces of elements in samples can also be determined quantitatively.
The disadvantage is that in the conventional and widespread "one-element search", only one chemical element is determined quantitatively at a time. A specific emission "lamp" is required for each element, with calibration prior to each test run. Once calibrated, hundreds of serial examinations are possible. But if no lamp is used for thallium , for example , then thallium in the sample will not be analyzed and not found, even if thallium might be present in the sample (if correctly stated, the analysis report would then say "Thallium: not examined" ). For example, to determine 10 heavy metals in a single sample, 10 lamp changes and 10 calibrations would be required. There are multi-element lamps for a number of elements. These can be provided with up to 3 elements without major problems. Otherwise, spectral interference can hardly be avoided. In addition, the elements have to match each other in terms of volatility. Nevertheless, traditionally the time and financial expenditure of this method for individual samples was relatively large. Considerable improvements have been made possible by continuous light sources (xenon discharge lamps), which cover all elements and largely eliminate the above-mentioned disadvantages.
Web links
Directory of databases and reference works with AAS spectra
literature
- Bernhard Welz , Michael Sperling: atomic absorption spectrometry. 4th edition. Weinheim 1999, ISBN 3-527-28305-6 .
- DA Skoog, JJ Leary: Instrumental Analytics. Springer, Berlin 1996, ISBN 3-540-60450-2 .
- DC Harris: Quantitative Chemical Analysis 7th Edition. WH Freeman and Company, New York 2003, ISBN 0-7167-7694-4 .
- K. Cammann: Instrumental Analytical Chemistry. Spektrum Akademischer Verlag, 2000, ISBN 3-8274-0057-0 .
- G. Wünsch: Optical analysis methods for the determination of inorganic substances. (= Göschen Collection. Volume 2606). Verlag de Gruyter, Berlin 1976, ISBN 3-11-003908-7 .
- RD Beaty, JD Kerber: Concepts, Instrumentation and Techniques in Atomic Absorption Spectrophotometry . 2nd Edition. The Perkin Elmer Corporation, 1988, OCLC 843073654 . (Book on AAS as a PDF document in English; 422 kB).
Individual evidence
- ↑ Bernhard Welz, Michael Sperling, Atomic Absorption Spectrometry, 3rd Ed., Wiley-VCH, Weinheim-New York, 1999, p. 107.