
hemoglobin
| Hemoglobin α subunit | ||
|---|---|---|

|
||
| The hemoglobin A 1 of adults consists of 2 α-chains (red) and 2 β-chains (blue) with 4 heme groups (green), each of which can bind an O 2 molecule (model according to PDB 1GZX ) | ||
|
Existing structural data : UniProt entry |
||
| Mass / length primary structure | 16 kDa per subunit; α chain 141, β chain 146 amino acids | |
| Cofactor | Hamm | |
| Identifier | ||
| Gene name (s) | HBA1 , HBA2 | |
| External IDs | ||
| Occurrence | ||
| Homology family | Beta-2 globin | |
| Parent taxon | Vertebrates | |
| Hemoglobin β subunit | ||
|---|---|---|

|
||
| Sphere model of the heme pocket of the hemoglobin β subunit with heme, iron (green) and dioxygen, according to PDB 1GZX | ||
|
Existing structural data : UniProt entry |
||
| Properties of human protein | ||
| Mass / length primary structure | 146 amino acids | |
| Cofactor | Hamm | |
| Identifier | ||
| Gene name | HBB | |
| External IDs | ||
| Occurrence | ||
| Homology family | Beta-2 globin | |
| Parent taxon | Vertebrates | |
| Parent |
| Cytosol |
| Gene Ontology |
|---|
| QuickGO |
Hemoglobin (from ancient Greek αἷμα haíma , "blood", and Latin globus , "lump, ball"), abbreviation Hb , is the iron-containing protein complex that is contained as a blood pigment in the red blood cells of vertebrates, binds oxygen and transports it in the bloodstream .
The hemoglobin of mammals is a tetramer , it consists of four globins as subunits . In adults, there are two Hb α and Hb β in hemoglobin A (Hb A 1 ), the most common form. The four proteins forming the complex are amino acid chains (α-chain 141 AA ; β-chain 146 AA) in the fold typical for globins, each with a pocket in which an iron (II) complex , the heme , is bound. Its iron ion is able to bind an oxygen molecule. The color of the heme changes from dark to light red. The strength of the bond is sensitive to the conformation of the protein environment of the heme. Interactions between the four globins favor the two extreme states in which the entire complex is either saturated with four molecules of oxygen (in the lungs or the gills ) or has given up all oxygen. Interactions with other molecules support both loading and unloading.
For information on the occurrence of hemoglobin and similar globins in the animal kingdom, see oxygen transporters .
history
The oxygen transport protein hemoglobin was discovered by Friedrich Ludwig Hünefeld in 1840 . In 1851 Otto Funke described the crystallization of hemoglobin by diluting animal blood with water, ethanol or diethyl ether and then slowly evaporating the solvent from the protein solution obtained ("Funkesche Kristalle"). The reversible oxygenation of hemoglobin was first reported by Felix Hoppe-Seyler in 1866 . The name hemoglobin also comes from him. The structural formula of heme (or the corresponding heme), i.e. the iron-containing porphyrin complex, was formulated by the German chemist William Küster as early as 1912 , and the chemist Hans Fischer was able to prove the correctness of this structural formula in 1928 through the complete synthesis of the heme. In 1930 he was awarded the Nobel Prize for Chemistry for this. Hemoglobin is one of the best-studied proteins; its structure was one of the first to be determined by Max Perutz et al in 1959 with the help of X-ray crystallography . For this work he and John Kendrew received the Nobel Prize in Chemistry in 1962 .
structure
Unmodified basic structure
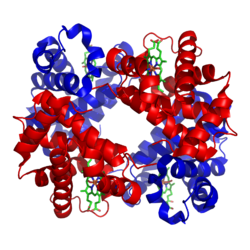
Ribbon model of hemoglobin A (HbA) -
two subunits α and β (red and blue) each carry a heme (rod model, green) as a prosthetic group (according to PDB 1GZX )
Structural formula of heme b

Mammalian hemoglobins consist of four subunits , two of the α and two of the β type. In each of these subunits a prosthetic group is embedded, where the oxygen binding takes place. One hemoglobin complex molecule can therefore bind four oxygen molecules. The prosthetic group of the oxygen-free form is an iron (II) complex of protoporphyrin IX, which occupies the four equatorial positions of the iron ion. The iron ion is in a high-spin state and is therefore a little too large to fit into the porphyrin's hole. So it is a little below the level of the ring. This heme b is bound to the protein matrix via the axial position of the iron ion on the underside via a proximal histidine residue. The second axial position on the upper side is unoccupied and is available for the attachment of the oxygen molecule.
Post-translational modifications
In addition to various rare modifications of individual amino acids in the hemoglobin subunits of humans, glycation of both subunits on special amino acids often occurs . This is the result of a high glucose concentration in the blood and can therefore be used in laboratory diagnostics to determine the average blood sugar level over the last few months.
During the glycation of hemoglobin, glucose becomes covalently attached to lysine -8, -17, -41, -62 of the α-subunit, and valine -2, lysine-9, -18, -67, -121 or -145 of the β-subunit bound. If a glycated β-hemoglobin is modified at valine-2 and the glucose residue has been converted to a stable ketoamine via an aldimine and an Amadori rearrangement , it is referred to as HbA1c .
Oxygen transport
Efficiency

Hemoglobin is a globular protein with very good solubility in water (solubility up to 5 mmol / l hemoglobin (34%)). 1 g of Hb can bind 1.389 ml of oxygen in vitro , but only 1.34 ml in vivo ( Hüfner's number ), so 100 ml of blood containing around 15 g of Hb can hold up to 15 × 1.34 ml at 100 percent saturation = Absorb 20.1 ml of oxygen.
The sigmoidal (S-shaped) course of the binding curve is striking. Normally, one would expect that the oxygen load would initially increase sharply and then more slowly with increasing oxygen partial pressure, as with myoglobin (hyperbolic course). For hemoglobin, the oxygen binding curve is unusually flat in the area of the oxygen partial pressure prevailing in the lungs and unusually steep in the area of the oxygen partial pressure prevailing in the tissue. The flat course of the binding curve in the end section prevents a greater decrease in oxygen saturation in old age, with lung dysfunction and at high altitudes, and the steeper course in the middle section ensures that a lot of oxygen is released when the venous oxygen partial pressure drops.
Normal range
A normal range has been established for the so-called “adult hemoglobin” (see below), which is predominant in adults. Other standard values apply to children.
The normal range is the range in which the Hb values of 96 percent of all healthy people are.
| g / dl (old unit) |
mmol / l ( SI unit ) |
|
|---|---|---|
| Men | 13.5-17.5 | 8.4-10.9 |
| Women | 12-16 | 7.4-9.9 |
| Newborn | 19th | 11.8 |
An increased hemoglobin value usually also means an increased number of erythrocytes ( polyglobulia ). B. occur when staying at high altitudes (lack of oxygen) or due to fluid loss. If the cause is unclear, it must also be clarified whether the significantly increased values are due to the disease polycythemia vera .
A decreased hemoglobin value is called anemia , a decreased load of the red blood cells with hemoglobin is called hypochromasia .
An increased / decreased hemoglobin value always depends on the normal value. If the normal value is 10.9 mmol / l, a value of 8.4 mmol / l can lead to symptoms of anemia . If the normal value is 9 mmol / l, no symptoms occur at 8.4 mmol / l.
The level of the hemoglobin value is decisive for the approval for blood donation . Men must have a minimum value of 8.4 mmol / l (13.5 g / dl), women one of 7.8 mmol / l (12.5 g / dl) in order to be approved by the donor doctor. The Hb value is determined using an electronically measuring Hb photometer . Current information from the DRK blood donation service states that men with an Hb of> 11.2 mmol / l (18.0 g / dl) are no longer allowed to donate (12/2006). However, this is not the case in all federal states; if the Hb level is elevated, further fluid intake is recommended before the donation.
The determination of the hemoglobin content is based on the detection of the heme groups of the protein. Therefore, the molar hemoglobin concentration is traditionally given as the concentration of the individual subunits (mean molar mass : 16114.5 g / mol). The conversion factor from g / dl to mmol / l is therefore 0.6206. According to IUPAC and DIN 58931, the (monomeric) hemoglobin concentration calculated in this way is referred to as Hb (Fe) . Since a hemoglobin complex molecule consists of 4 subunits, each with a heme group, the values given in mmol / l must be divided by 4 in order to obtain the concentration of the hemoglobin tetramer (referred to as Hb according to IUPAC and DIN 58931 ) (molar mass: 64458 g / mol). The conversion factor from g / dl to mmol / l is therefore 0.1551. The values given in g / dl remain unchanged.
Oxygen binding through hemoglobins at the molecular level


When oxygen is bound, a dioxygen (O 2 ) molecule is absorbed into the center of the heme complex. The atom of the central iron ion (Fe II ) changes into a low-spin state due to the oxygen bond. In the process, its size is reduced and it slips into the plane of the porphyrin ring.
The bound oxygen molecule is stabilized via a hydrogen bridge . This is formed with the side chain of the globin protein : a distal histidine residue that is located near the central atom. The proximal histidine residue, via which the iron atom of the heme is bound to the protein matrix, is on the other side of the plane of the ring.
Cooperative effect on oxygen binding
A hemoglobin, which consists of four Hb subunits, can bind four oxygen molecules. For purely statistical considerations, it would be expected that the effort to bind further oxygen molecules would decrease with each oxygen molecule that is already bound. However, studies have shown that the opposite is the case and that the oxygen affinity increases with increasing load (positive cooperativity ).
pH value dependence of oxygen binding
The equilibrium between the R and T forms is pH-dependent ( Bohr effect ) and is shifted at a low pH value by protonation in favor of the less oxygen-affine T-form. The same effect has a high CO 2 - partial pressure by a reversible carboxylation of the subunits. This leads to high proton and carbon dioxide concentrations, such as those found in e.g. B. prevail through cellular respiration and lactic acid fermentation in the working muscle, favor a complete discharge of the hemoglobin.
Competition between oxygen and carbon monoxide
Carbon monoxide (CO) is very poisonous because it competes with oxygen for the axial coordination points of the iron centers. Once bound, CO can e.g. B. can only be displaced by oxygen pressure treatment in a pressure chamber - it thus blocks binding sites for oxygen. In heavy smokers, up to 10 percent of the hemoglobin is occupied by CO.
Carbon monoxide binds 25,000 times more strongly to
free heme b than dioxygen, but only about 200 times in hemoglobin. The reason for the reduced CO affinity is that due to the space required by the distal histidine, the linear Fe-CO coordination preferred by carbon monoxide is not possible.
Effects that affect oxygen binding
The oxygen binding curve is shifted to the right on the abscissa by:
- Temperature rise
- Drop in pH
- Increase in the concentration of 2,3-bisphosphoglycerate in the erythrocytes
- Increase in the concentration of carbon dioxide
The shift to the right makes the hemoglobin more easily releasing oxygen . An example: a working muscle needs a lot of oxygen to contract . Since it converts some of the energy into heat, the temperature rises in the working muscles. It also releases lactic acid and the pH value drops. The increased metabolism produces more carbon dioxide: the local effects allow the muscles to take more oxygen from the blood.
- The muscles have myoglobin (see below), which has a higher affinity for oxygen (it attracts more oxygen). It serves as an oxygen store.
The oxygen binding curve is shifted to the left on the abscissa by:
- Temperature decrease
- Increase in pH
- Decrease in the concentration of 2,3-bisphosphoglycerate in the erythrocytes
- Decrease in the concentration of carbon dioxide
The left shift causes hemoglobin to bind oxygen more tightly. This is done z. B. in cardiac surgery by hypothermia to saturate his blood maximally with oxygen. Animals that hibernate also benefit from this effect. In the lungs, part of the carbon dioxide is released in the course of exhalation - the Hb can be reloaded with oxygen more easily.
The 2,3-bisphosphoglycerate formed by the enzyme bisphosphoglycerate mutase in the Rapoport-Luebering cycle (bypass route to glycolysis) is the most important intermediate in glycolysis in red blood cells. It binds to hemoglobin and causes an allosteric effect ; the decrease in the affinity of hemoglobin for oxygen. It is vital to enable the release of oxygen in the organism. Under physiological conditions, 2,3-BPG is found in red blood cells in about the same concentration as hemoglobin. An increase in the 2,3-BPG concentration is e.g. B. to be observed during height adjustment. The purpose of this regulation is as follows: If the oxygen saturation in the blood is reduced by the “thin air” at high altitude, Hb releases the bound oxygen to the consumer more poorly than with high saturation (see binding curve). Nevertheless, the supply of all organs with O 2 must be guaranteed. So Hb has to become less affine for oxygen in order to adequately supply the periphery .
Hemoglobin types
Different hemoglobins can be found in humans in different phases of life (embryonic phase, fetal phase and after birth, i.e. the “adult phase”). These hemoglobins differ in their affinity for oxygen. They each consist of four subunits in different combinations in pairs.

Historically, the different subunits of hemoglobin were created by duplicating the globin gene. The combinations of these subunits as tetramers are synthesized at different times depending on the oxygen requirement , for example in order to obtain oxygen from the maternal blood as a fetus in the womb.
During pregnancy , oxygen is transported through the placenta ( placenta ) to the fetus , which it then takes up efficiently. This is ensured by the fact that the embryonic and fetal hemoglobin types have a significantly higher oxygen-binding affinity than the adult hemoglobin formed later after birth. In addition, the hematocrit is greatly increased compared to the mother. This allows enough oxygen to reach the fetus through the umbilical cord.
Embryonic hemoglobins
The embryonic hemoglobins are formed in the embryonic phase, the first 8 weeks after fertilization, in blood islands of the yolk sac and have proper names:
- Gower-1 (ζ 2 ε 2 ) ("zeta-epsilon")
- Gower-2 (α 2 ε 2 ) ("alpha-epsilon")
- Portland-1 (ζ 2 γ 2 ) ("zeta-gamma")
- Portland-2 (ζ 2 β 2 ) ("zeta-beta")
Fetal hemoglobins
As fetal hemoglobin , hemoglobin F (HbF) mainly formed during the fetal period (from the 9th week after conception to birth) is called. The synthesis of fetal hemoglobin begins in the previous embryonic phase and is not stopped immediately after birth, but continues for a few months. The place of formation are the liver and spleen . It has a much higher affinity for oxygen than adult hemoglobin to take in the oxygen from the maternal blood.
- Hemoglobin F (α 2 γ 2 ) - The dominant hemoglobin in the fetus, only detectable in traces in healthy adults
Adult hemoglobins
As adult haemoglobins hemoglobin A valid 1 (HbA 1 ) (Syn. Hemoglobin A (HbA)) and hemoglobin A 2 (HbA 2 ). The synthesis of adult hemoglobins begins in the fetus and then replaces fetal hemoglobin in the first few months after birth. The place of education is the bone marrow . During the first year of life, the expression of the γ gene is lowered and that of the β gene is increased. The transcription factor BCL11A is the decisive repressor of γ-globin in adults. Therefore, fetal hemoglobin is only detectable in traces in adults.
- Hemoglobin A 1 (α 2 β 2 ) - 98%
- Hemoglobin A 2 (α 2 δ 2 ) - 2%.
Glycohemoglobins
Glycohemoglobins are formed by binding glucose to amino groups of the globins. Under physiological conditions, this happens non-enzymatically through the contact of the hemoglobin with the blood sugar ( glycation ). The main component HbA 1 of hemoglobin A is mainly converted into HbA 1c via the intermediate stages HbA 1a and HbA 1b , the proportion of which therefore represents the average blood sugar level over the past few weeks.
Methemoglobin
Methemoglobin (Met-Hb) is a deactivated, non-oxygen-affine form of hemoglobin in which the iron ion is in oxidation level III instead of II. The NADH -dependent enzyme methemoglobin reductase (diaphorase I) is able to convert methemoglobin back into hemoglobin. Typically, two percent of human hemoglobin is methemoglobin. A higher proportion can either be genetic or the result of poisoning. A high proportion of methemoglobin results in an inadequate supply of oxygen to the organism.
Breakdown of hemoglobin
When the red blood cells have reached the end of their life (around 120 days), they are broken down in the mononuclear phagocytes mainly in the spleen (and in the liver and bone marrow when there is a high level of degradable hemoglobin). The breakdown process begins in the spleen and continues in the liver. First the globin part is separated from the heme and degraded to amino acids. The heme is split into biliverdin via a cytochrome P450-dependent oxygenase ( hemooxygenase ), with iron (Fe 2+ ) and carbon monoxide being released. The iron is released from the macrophages to the transport protein transferrin , which is present in the blood . The biliverdin reductase finally converts the greenish biliverdin into bilirubin . This is released into the blood and coupled to albumin (because it is poorly soluble in water) and transported to the liver. Here the bilirubin is conjugated twice with glucuronic acid and thus made soluble - it can now be released into the intestine with the bile and excreted in the stool. In the intestine, bacteria ensure that the glucuronic acid is partially broken down again, and in this way the orange bilirubin is reduced to colorless urobilinogen and brown sterkobilinogen . A small proportion of this reduced bilirubin is taken up again via the intestine (enterohepatic circulation) and excreted via the kidneys (yellow urine ). Various liver diseases such as inflammation of the liver ( hepatitis ) or obstruction of the drainage in the gallbladder (gallstones) lead to an increased bilirubin concentration. Bilirubin is the pigment that, when deposited in the skin, leads to so-called jaundice (jaundice = yellowing of the skin and dermis ( sclera ) of the eyes).
Role in disease
Mutations in HBA1 - gene can be defects in the α-subunit and this to Heinz bodies - anemia and α-thalassemia lead. Mutations in the HBB gene can cause Heinz body anemia, β-thalassemia, and sickle cell anemia . Some variants of the HBG1 / HBG2 genes can cause neonatal jaundice .
Decreased hemoglobin levels, with or without a reduction in red blood cell counts, lead to the symptoms of anemia . There are many causes of anemia, with iron deficiency being the most common cause in the western world. Heme synthesis is inhibited by iron deficiency. As a result, the red blood cells are hypochromic (devoid of the red color) and microcyic (smaller than normal).
In a hemolysis (increased degradation of red blood cells) enters jaundice on, caused by the hemoglobin metabolite bilirubin . Up to an amount of 135 mg / dl, the hemoglobin released in the blood vessels is bound to haptoglobin ; if the blood falls heavily, free hemoglobin appears in the blood ( hemoglobinemia ).
A group of genetic defects known as porphyrias disrupt the synthesis of heme. The accumulation of heme precursors leads to photosensitivity, abdominal pain and neurological problems as well as porphyrinuria.
In methemoglobinemia, the hemoglobin present in the red blood cells, which is used to transport oxygen, is converted into the non-functional methemoglobin and is therefore no longer available for oxygen transport. The cause can be hereditary ( congenital methaemoglobinemia ) or it can be triggered by toxins.
Mutations in the globin chains are associated with various hemoglobinopathies , such as sickle cell anemia and thalassemia .
Malaria pathogens break down hemoglobin in red blood cells they have infected in order to produce proteins for their own metabolism. The heme creates hemozoin , which is crystallized by the parasite and can be seen as a pigment in the infected erythrocytes under the microscope . The malaria drug chloroquine inhibits this crystallization and the parasite is poisoned by the heme.
proof
The detection of hemoglobin is carried out by the Teichmann test, in which the blood is carefully warmed up with table salt and glacial acetic acid , whereby hemin (Teichmann crystals) are deposited, or with the luminol reaction , in which a solution of luminol and sodium hydroxide and a solution of hydrogen peroxide are used is used. This reaction only takes place in the presence of a catalyst; if hemoglobin is detected, this would be the iron (II) ion in the heme complex.
Hemoglobin in Art and Music

- In 2005, the German-American artist Julian Voss-Andreae created a sculpture based on the structure of hemoglobin. The intended rusting of the work is an allusion to the oxygenation in the heme.
- British rock band Placebo recorded a song called Hemoglobin .
- French rapper MC Solaar released a successful single in 1994 called La concubine de l'hémoglobine .
- The German melodic death metal band Deadlock has a song called "Killing The Time With Haemoglobin" with a length of 11 minutes on their album "The Arrival".
Individual evidence
- ^ Friedrich Ludwig Hünefeld: Chemism in the animal organization. Brockhaus, 1840 ( limited preview in Google Book Search).
- ↑ A NASA Recipe For Protein Crystallography (PDF; 782 kB) In: Educational Brief . National Aeronautics and Space Administration. Retrieved February 2, 2016.
- ↑ Hoppe-Seyler F: About the oxidation in living blood . In: Med-chem Exam Lab . 1, 1866, pp. 133-140.
- ^ Georg Hoppe-Seyler: Felix Hoppe-Seyler - doctor and natural scientist .
- ^ William Küster, in: Hoppe-Seylers Zeitschrift für Physiologische Chemie . No. 82, 1912, p. 463 ff.
- ↑ MF Perutz, MG Rossmann, AF Cullis, H. Muirhead, G. Will, ACT North: Structure of H . In: Nature . 185, No. 4711, 1960, pp. 416-422. doi : 10.1038 / 185416a0 . PMID 18990801 .
- ^ Perutz MF: Structure of hemoglobin . In: Brookhaven symposia in biology . November 13, 1960, ISSN 0068-2799 , pp. 165-83. PMID 13734651 .
- ↑ Perutz MF, Muirhead H, Cox JM, et al : Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: (1) x-ray analysis . In: Nature . 219, No. 5149, July 1968, pp. 29-32. doi : 10.1038 / 219029a0 . PMID 5659617 .
- ↑ Perutz MF, Muirhead H, Cox JM, Goaman LC: Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: the atomic model . In: Nature . 219, No. 5150, July 1968, pp. 131-9. PMID 5659637 .
- ↑ Canterbury Scientific: HbA1c and Glycated Hemoglobin ( Memento of the original from January 29, 2012 in the Internet Archive ) Info: The archive link has been inserted automatically and has not yet been checked. Please check the original and archive link according to the instructions and then remove this notice.
- ↑ UniProt P69905 , UniProt P68871
- ↑ JM Berg, JL Tymoczko, L. Stryer: Biochemistry. 6th edition. Spectrum Academic Publishing House, Elsevier GmbH, Munich 2007; P. 208ff; ISBN 978-3-8274-1800-5
- ↑ Robert F. Schmidt, Gerhard Thews (Ed.): Physiology of humans . 25th edition. Springer-Verlag, Berlin 1993, ISBN 3-541-02636-7 , pp. 616-620 .
- ↑ Erich Schütz, Heinz Caspers, Erwin-Josef Speckmann (eds.): Physiology . 16th edition. Urban & Schwarzberg, Munich 1982, ISBN 3-540-57104-3 , pp. 86-87 .
- ↑ a b c d R. Zander, W. Lang, P. Lodemann: The molecular weight of hemoglobin . Retrieved August 13, 2013.
- ↑ Adair GS: The hemoglobin system. VI. The oxygen dissociation curve of hemoglobin. In: J Biol Chem 1925; 63: 529-545.
- ↑ DE Koshland, G. Némethy, D. Filmer: Comparison of experimental binding data and theoretical models in proteins containing subunits. In: Biochemistry . Volume 5, Number 1, January 1966, pp. 365-385, ISSN 0006-2960 . PMID 5938952 .
- ↑ Hemoglobinopathies in the Fetal Position | NEJM. Accessed January 31, 2019 .
- ^ Vijay G. Sankaran, Tobias F. Menne, Jian Xu, Thomas E. Akie, Guillaume Lettre: Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A . In: Science (New York, NY) . tape 322 , no. 5909 , December 19, 2008, ISSN 1095-9203 , p. 1839–1842 , doi : 10.1126 / science.1165409 , PMID 19056937 .
- ↑ UniProt P69905 , UniProt P68871 , UniProt P69891
- ↑ Wolfgang Piper: Internal medicine . 2nd Edition. Springer, Berlin 2002, ISBN 978-3-642-33108-4 , pp. 639-640 .
- ↑ Constance Holden: Blood and Steel . (pdf) In: Science . 309, No. 5744, September 30, 2005, p. 2160. doi : 10.1126 / science.309.5744.2160d .
- ↑ Moran L, Horton RA, Scrimgeour G, Perry M: Principles of Biochemistry . Pearson, Boston, MA 2011, ISBN 0-321-70733-8 , p. 127.
literature
- MF Perutz: Stereochemistry of cooperative effects in hemoglobin. In: Nature , Volume 228, Number 5273, November 1970, pp. 726-739, ISSN 0028-0836 . PMID 5528785 .
- L. Makowski, J. Bardhan, et al. a .: WAXS studies of the structural diversity of hemoglobin in solution. In: Journal of Molecular Biology , Volume 408, Number 5, May 2011, pp. 909-921, ISSN 1089-8638 . doi: 10.1016 / j.jmb.2011.02.062 . PMID 21420976 . PMC 3081904 (free full text).
- T. Yonetani, M. Laberge: Protein dynamics explain the allosteric behaviors of hemoglobin. In: Biochimica et biophysica acta , Volume 1784, Number 9, September 2008, pp. 1146-1158. doi: 10.1016 / j.bbapap.2008.04.025 . PMID 18519045 . PMC 2668241 (free full text). (Review).
- Martin D. Vesper, Bert L. de Groot, Dennis R. Livesay: Collective Dynamics Underlying Allosteric Transitions in Hemoglobin. In: PLoS Computational Biology , Volume 9, 2013, p. E1003232, doi: 10.1371 / journal.pcbi.1003232 .
Web links
- Interactive 3D Model of Hemoglobin - Proteopedia
- Molecule of the month: hemoglobin (english)
- Jennifer McDowall / Interpro: Protein Of The Month: Hemoglobin. (engl.)