Bauchspeicheldrüsenkrebs

Pankreastumoren sind Tumoren der Bauchspeicheldrüse (griech.: πάγκρεας, pánkreas). Die Mehrheit der Pankreastumoren ist bösartig und befällt den die Verdauungsenzyme bildenden Anteil der Bauchspeicheldrüse, und hier vorwiegend die Gänge innerhalb des Organs. Diese „duktalen Adenokarzinome“ gehören zu den häufigen Krebserkrankungen. Die wichtigsten Risikofaktoren sind Bauchspeicheldrüsenentzündungen, Zigarettenrauchen und einige Erbkrankheiten. Häufigste Symptome sind eine stetig zunehmende Gelbsucht, in den Rücken ausstrahlende und nachts an Stärke zunehmende Bauchschmerzen sowie Verdauungsstörungen und Gewichtsverlust.
Pankreaskarzinome wachsen aggressiv und bilden früh Tochtergeschwulste (Metastasen). Sie können bisher nur durch operative Entfernung behandelt werden, allerdings werden vier von fünf bösartigen Pankreastumoren erst in einem so weit fortgeschrittenen Stadium erkannt, dass keine Heilung mehr möglich ist. Chemo- und Strahlentherapie haben in der Behandlung von Pankreastumoren nur eine unterstützende Funktion. Bei der Operation wird die Bauchspeicheldrüse teilweise oder vollständig entfernt (Pankreatektomie) und die unterbrochenen Gallen- und Pankreasgänge wieder mit dem Darmtrakt verbunden. Obwohl die Heilungsrate gegenüber früheren Jahrzehnten verbessert werden konnte, nehmen Palliation (Linderung) und bestmöglich unterstützende Behandlungsmaßnahmen (Best Supportive Care) in den Pankreaszentren immer noch breiten Raum ein.
Geschichte
Der griechische Arzt Rufus von Ephesos soll die Bauchspeicheldrüse erstmals als Organ beschrieben haben. Galenos bemerkte, dass sie ein Sekret absondert, hielt das Organ aber für ein Polster des Magens – eine Vorstellung die sich bis Mitte des 17. Jahrhunderts hielt.[1] Die Verdauungsfunktion des Sekrets wurde erst Mitte des 19. Jahrhunderts von Claude Bernard endgültig bewiesen.[2]
Operationen der Bauchspeicheldrüse, vor allem mit Entfernung (Resektion) größerer Anteile der Drüse, galten lange als unmöglich. Erst gegen Ende des 19. Jahrhunderts gelangen überhaupt einzelne derartige Eingriffe, und bis 1898 waren erst neun Entfernungen veröffentlicht worden. Bekannt ist eine erfolgreiche Linksresektion (s. u.) durch Friedrich Trendelenburg 1882. 1890 entdeckten Oskar Minkowski und Josef von Mering, dass die Entfernung der Bauchspeicheldrüse die Zuckerkrankheit verursacht und damit indirekt das Insulin,[3] welches dann 1921 von Frederick Banting und Charles Best erstmals isoliert wurde[4]. Im Jahr 1940 standardisierte Allen Oldfather Whipple die von Walther Kausch und vermutlich auch etwa gleichzeitig von Alessandro Codivilla entwickelte[5] Operationstechnik, bei der Bauchspeicheldrüse und Zwölffingerdarm gemeinsam entfernt werden. Diese Vorgehensweise zählt bis heute zu den gebräuchlichen Standardverfahren. Das schwierigste Problem der Resektionen bestand stets darin, den Abfluss des Bauchspeicheldrüsensekrets in den Darm sicherzustellen. Ab etwa 1950 erarbeiteten Chirurgen dafür verbesserte Methoden und Instrumente, beginnend mit der Idee von Charles Frey, den Rest der Bauchspeicheldrüse mit einer eröffneten Darmschlinge zu vernähen.
Anatomie

Die Bauchspeicheldrüse ist eine 40 bis 120 g schwere Drüse im Retroperitoneum, hinter dem Magen, links vom Zwölffingerdarm und wird in einen Kopf, einen Körper und einen Schwanz untergliedert. Der Schwanz berührt links Milz und Niere. Der Hauptgallengang durchquert rechts den Kopf der Bauchspeicheldrüse. Die Aorta und mehrere große Eingeweidearterien (Truncus coeliacus, obere Gekrösearterie, Milzarterie) haben engen Kontakt zur Bauchspeicheldrüse, ebenso die untere Hohlvene und die Pfortader.[6] Alle diese Strukturen können von Pankreastumoren erfasst werden. Die Ausführungsgänge für das Pankreassekret (Wirsung- und der beim Menschen nur selten vorkommende Santorini-Gang) verlaufen durch das ganze Organ und können vom Tumor verstopft werden. Seine Blutversorgung erhält das Pankreas aus dem Truncus coeliacus und der oberen Gekrösearterie, manchmal auch aus einer abweichenden Leberarterie, was Operationen erheblich erschweren kann. Das verbrauchte Blut fließt in die obere Gekrösevene und in die Milzvene ab. Die Lymphe strömt in alle Richtungen zu den nächstgelegenen Lymphknotengruppen (Bauchspeicheldrüsen-, obere und untere Bauchspeicheldrüsen-Zwölffingerdarm-, Leber-, Zölikial-, obere Mesenterial-, Milz- und obere paraaortale Lymphknoten), die alle von einer Metastasierung betroffen sein können.[7]
Die Bauchspeicheldrüse ist überwiegend exokrin („nach außen ausscheidend“): Ihr Verdauungssekret wird über die beiden Ausführungsgänge in den Zwölffingerdarm abgegeben. Sie enthält aber auch endokrine („nach innen ausscheidende“) Zellgruppen in den sogenannten Langerhans-Inseln, die die Hormone (unter anderen Insulin und Glucagon) produzieren und an das Blut abgeben. Beide Gewebe können prinzipiell Tumoren entwickeln. Über 95 % der Pankreastumoren entstehen aus dem exokrinen Organ, genauer gesagt aus dem Gangepithel und den Azinuszellen.[8] Neben den gutartigen Zystadenome und muzinösen Zystomen sind es vor allem Karzinome. Tumoren des endokrinen Pankreasgewebes kommen dagegen fast nur bei erblichen Syndromen vor. Sie werden zu den neuroendokrinen Tumoren gerechnet.
Drei Viertel der Tumoren entstehen im Pankreaskopf, dem am weitesten rechts, am Zwölffingerdarm gelegenen Anteil. 20 % der Tumoren treten im mittleren Anteil (Corpus) und fünf Prozent im linksseitigen Pankreasschwanz auf.
Häufigkeit und Ursachen

Das Pankreaskarzinom, der mit Abstand häufigste Pankreastumor, verursacht in Deutschland etwa 14.000 Neuerkrankungen jährlich – die Neuerkrankungsrate liegt bei rund 18 pro 100.000 Einwohner und Jahr.[9] In der deutschen Krebsstatistik steht es bei Männern an der zehnten, bei Frauen an neunter Stelle (Stand 2006). Bei den Krebstodesfällen belegt es den vierten Platz.[10] Die Kranken sind meist über 60 Jahre alt und häufiger Männer als Frauen (1,6:1).
Wichtigster Risikofaktor neben dem Lebensalter ist die chronische Pankreatitis (Bauchspeicheldrüsenentzündung): Etwa einer von 50 davon Betroffenen entwickelt innerhalb von zehn Jahren ein Karzinom. Diabetes verdoppelt das Karzinomrisiko aus bislang unbekanntem Grund. Lebensstilbezogene Gefahren sind Zigarettenrauchen (Raucherentwöhnung reduziert das Risiko nach zwei Jahren auf die Hälfte), Vitamin-D-Mangel, starkes Übergewicht (BMI > 30) und fettreiche Ernährung.[11] Angeborene Risiken sind Syndrome mit allgemein gesteigerter Krebserkrankungsrate wie das Peutz-Jeghers-Syndrom, die erbliche Pankreatitis und die zystische Fibrose. Zwei oder mehr Pankreaskarzinomfälle in der nahen Verwandtschaft erhöhen das Risiko um ein Vielfaches. Screeninguntersuchungen werden gegenwärtig nur für Familien mit erblicher Pankreatitis oder mehreren Pankreaskarzinomfällen empfohlen; üblich ist dann eine jährliche Endosonografie ab dem 50. Lebensjahr.[12]
Chemische Karzinogene (Naphthylamin, Benzidin oder Nitrosamine) können das Erkrankungsrisiko erhöhen. Auch chlororganische Verbindungen und polycyclische aromatische Kohlenwasserstoffe werden verdächtigt.[13]
Für die Karzinome sind ein aggressives Wachstum, eine schnelle Metastasierung und ein schlechtes Ansprechen auf die verfügbaren Behandlungen kennzeichnend. Das liegt auch an dem besonders hohen Entartungsgrad: wachstumsregulierende oder tumorunterdrückende Gene wie jene für HER2/neu, KRAS, p16, p53 und DPC4 sind in den Tumorzellen sehr häufig durch Mutationen inaktiviert.[14]
Pathologie

_Case_01.jpg)
Pankreastumorzellen können Ähnlichkeit mit Zellen der Gänge (duktal), der Azini und Langerhans-Inseln aufweisen, aber auch gemischten Charakter haben.[15] In der Regel richten sich die Pathologen nach der WHO-Klassifikation bösartiger Tumoren, derzeit in der Ausgabe von 2010.[16] Die meisten bösartigen Tumoren (Malignome) werden danach als Varianten des duktalen Adenokarzinoms bezeichnet, eingestuft von hochdifferenziert bis undifferenziert. Dazu kommen Azinuszellkarzinome, muzinöse Zystadenokarzinome und intraduktale Neoplasien (s. u.). Endokrine Langerhanszell-Karzinome, nichtepitheliale Malignome (Lymphome und Sarkome) sind selten, ebenso die Pankreasmetastasen von anderen Organtumoren. Der häufigste gutartige Tumor ist das seröse Zystadenom.
Adenokarzinom
Adenokarzinome des Gangsystems können sich direkt bilden oder aus sogenannten Präkanzerosen entwickeln. Dies sind oberflächliche Wucherungen des Epithels, wobei vor allem papilläre Hyperplasien – nach neuer Nomenklatur pankreatische epitheliale Neoplasien 3 (PanIN 3) – als gefährlich gelten. Gutartige Tumoren wie das Zystadenom und die intraduktalen papillären Neoplasien weisen mit zunehmender Größe ebenfalls ein zunehmendes Entartungsrisiko auf.
Bei den Adenokarzinomen kann man im Mikroskop je nach ihrem Entartungsgrad noch schleimgefüllte Drüsenschläuche mit Zylinderepithel erkennen („duktaler Typ“). Die Nervenscheiden sind fast immer tumorinfiltriert. Charakteristisch ist außerdem eine Verdichtung des umgebenden Bindegewebes („desmoplastische Reaktion“), die in den bildgebenden Verfahren schlecht vom eigentlichen Tumor zu unterscheiden ist. Die wichtigsten histologischen Varianten des duktalen Adenokarzinoms sind das adenosquamöse Karzinom, das muzinöse nichtzystische Karzinom und das anaplastische (undifferenzierte) Karzinom. Nach dem Grad der Entdifferenzierung vergibt der Pathologe das Grading G1 bis G4.
Karzinome sind bei Diagnosestellung meist zwei bis fünf Zentimeter groß (bildgebend nachweisbar ab etwa einem Zentimeter Größe). Sie sind unscharf begrenzt, von fester Konsistenz und grau-gelblicher Farbe, oft zentral nekrotisch. Es kommt häufig zu einer Verengung (Stenose) der durch die Bauchspeicheldrüse verlaufenden Strecke des Gallengangs, häufig auch zu einer Stenose des Pankreas-Ausführungsgangs. Der Tumor kann in die Wand des Zwölffingerdarmes einwachsen, weiterhin auch wichtige Gefäßstrukturen wie die obere Gekrösearterie, die Milzvene, die Pfortader und/oder die untere Hohlvene infiltrieren. Die Feststellung dieser Beteiligungen ist für die Stadienbestimmung (Staging) und damit für das weitere therapeutische Vorgehen von großer Bedeutung.
Die meisten duktalen Adenokarzinome exprimieren die Mucine 1 (Ca 15-3), 3, 5/6, und 16 (Ca 125) sowie das Glykoprotein CA 19-9 auf den Zellmembranen.
Die ersten Metastasen finden sich in den benachbarten Lymphknoten und – über den Blutstrom der Pfortader – in der Leber. Tumoren im Pankreaskörper und Pankreasschwanz sind bei Diagnosestellung zumeist größer als Pankreaskopftumoren und haben meistens schon zu Lebermetastasen oder einer Infiltration des Bauchfells (Peritonealkarzinose) geführt.
Tumoren der Papilla Vateri
Die Tumoren im Bereich der gemeinsamen Mündung des Gallen- und Pankreasgangs (Papilla Vateri) sind meist Adenokarzinome; sie sollen manchmal aus tubulovillösen Adenomen hervorgehen. Die Prognose des Papillenkarzinoms ist relativ gut, da die rasch auftretende Gelbsucht zu einer frühzeitigen Erkennung führt. Ausbreitung und Metastasierung verlaufen wie beim Pankreaskarzinom.
Intraduktaler papillär-muzinöser Tumor (IPMT)

Der IPMT wird auch als Intraduktales papillär-muzinöses Karzinom bezeichnet. Dieser Tumor breitet sich innerhalb des Gangsystems aus, beginnend meist im Kopfteil der Bauchspeicheldrüse. Man unterscheidet einen Hauptgangtyp (schlechtere Prognose), einen Seitenasttyp und einen kombinierten Typ. Das normale Gangepithel wird durch hochzylindrische neoplastische Zellen in kleinknotig-warzenförmigen (papillären) Wucherungen ersetzt, die viskösen Schleim bilden, der nur schwer abfließt und den Gangabschnitt unregelmäßig bis auf drei oder vier Zentimeter erweitert. Die Tumorzellen können die gesamte Bauchspeicheldrüse erfassen. Bei etwa 30 % der Patienten bestehen schon Gefäßeinbrüche und damit ein invasives Karzinom, wegen der Schleimseen im mikroskopischen Bild wird es oft als muzinöses nichtzystisches Karzinom oder Kolloidkarzinom bezeichnet. Dennoch ist die Prognose dieser Tumorart vergleichsweise sehr gut mit >90 % Langzeitüberleben.
Muzinös-zystischer Tumor
Der Muzinös-zystische Tumor wird auch muzinöses Zystadenom oder Zystadenokarzinom genannt. Dieser Tumor kann gut- oder bösartig sein. Computertomographie- oder Magnetresonanztomographie-Bilder können diese nicht unterscheiden, deshalb werden gut- und bösartige Varianten unter diesem Begriff zusammengefasst und unabhängig von den Symptomen immer operiert. 40- bis 60-jährige Frauen sind bevorzugt betroffen. Die 2 bis 12 cm großen Tumoren weisen eine breite Bindegewebskapsel auf. Sie bestehen meist aus weniger als sechs großen Zysten, die mit muzinproduzierendem Zylinderepithel ausgekleidet sind. Gelingt die operative Entfernung, ist die Prognose dieses Tumors gut; selbst die bösartige Variante erreicht Fünf-Jahres-Überlebensraten um 75 %.
Azinuszellkarzinom
Dieser seltene Tumor der Azinuszellen kommt doppelt so häufig bei Männern wie bei Frauen vor (Altersgipfel: 55–65 Jahre). Obwohl die Tumoren gewöhnlich relativ groß sind (4–6 cm), werden sie oft erst entdeckt, wenn sie bereits in die Leber metastasiert sind. Gelegentlich kommt es, durch eine massive Sekretion von Lipasen bedingt, zu Fettgewebsnekrosen unter der Haut sowie zu Gelenkschmerzen.
Seröses Zystadenom
Das Seröse Zystadenom, auch als mikrozystisches (Zyst-)Adenom bezeichnet, ist ein gutartiger Tumor, der vorwiegend bei Frauen im höheren Lebensalter beobachtet wird. Er liegt häufiger im Pankreaskopf, jedoch kann jede Region betroffen sein. Zystadenome können sechs bis zehn Zentimeter groß werden. Sie bestehen im Unterschied zum muzinös-zystischen Tumor (s. o.) aus kleinen Zysten mit serösem Inhalt, die durch zarte Septen getrennt sind. Im Zentrum findet man eine narbenartige Verdichtung und oft auch Verkalkungen. Diese Zysten sind mit kubischem Epithel ausgekleidet, histologisch finden sich keine Atypien oder Mitosefiguren. Eine Assoziation mit dem Von-Hippel-Lindau-Syndrom wurde beschrieben, der Tumor kann dabei große Abschnitte der Bauchspeicheldrüse einnehmen. Das seröse Zystadenom weist keine Entartungstendenz auf und sollte nur entfernt werden, wenn es durch seine Größe Symptome verursacht.
Endokrine Tumoren
Endokrine Pankreastumoren (Synonym pankreatische neuroendokrine Neoplasien PaNEN, veraltet: Karzinoide) entstehen aus den endokrinen Drüsenzellen der Bauchspeicheldrüse und bilden nur etwa 1–2 % der Pankreastumoren. Höchstens 50 % sind funktionell, d. h. sie bilden vermehrt Hormone und verursachen dadurch Krankheitserscheinungen.[17] Dazu zählen:
- Insulinom
- Gastrinom (Zollinger-Ellison-Syndrom)
- Somatostatinom
- Glucagonom
- VIPom (Verner-Morrison-Syndrom)
Ein gehäuftes Auftreten findet man beim Syndrom der multiplen endokrinen Neoplasie (MEN1-Syndrom). Davon abgesehen gibt es neuroendokrine Pankreastumoren im Kindesalter praktisch gar nicht; später treten sie in allen Altersklassen sowie bei Männern und Frauen etwa gleich selten auf. Die Prävalenz liegt unter 1/100000.
Endokrine Tumoren sind begrenzte, einzeln auftretende, runde Tumoren mit einem Durchmesser von 1 bis 4 cm, die in allen Teilen des Pankreas auftreten können. Histologisch handelt es sich um einheitlich aussehende Tumorzellen mit einem feinkörnigen Zytoplasma. Die Zellen sind solide, trabekulär und pseudoglandulär angeordnet. Immunhistologisch sind endokrine Tumoren positiv für die Marker NSE, Synaptophysin und Chromogranin A (CgA), letzterer ist auch im Blutserum bei vielen Erkrankten erhöht.[18] Im Elektronenmikroskop sieht man in den Tumorzellen neurosekretorische Hormongranula.
Nach der WHO-Klassifikation von 2010 sind alle PaNEN potentiell bösartig. Die histologisch gut differenzierten Tumoren (neuroendokrine Tumoren NET) werden nach dem Ki67/MIB1-Index in hoch- und mitteldifferenziert (<2 % = G1, 2–20 % = G2) unterteilt. Die hochproliferierenden (Ki67-Index > 20 %) sogenannten neuroendokrinen Karzinome NEC werden als G3 eingestuft und nochmals in klein- und großzellige Subtypen unterteilt.
Kriterien zur prognostischen Einschätzung von neuroendokrinen Pankreastumoren (WHO 2010) sind neben diesem Differenzierungsgrad und der TNM-Klassifikation noch das Vorliegen von mikroskopischen Gefäßeinbrüchen und die hormonelle Aktivität:
| Metastasierungsrisiko | Histologie | Differenzierung | TNM |
|---|---|---|---|
| minimal (benignes Verhalten) | NET, keine Angioinvasion | G1 | T1 N0 M0 |
| gering | NET | G2 | T1-2 N0 M0 |
| erheblich | NET | G1-2 | T2>4 cm oder T3, N0-1, M0-1 |
| hochmalignes Verhalten | NEC | G3, funktionell inaktiv | jedes T, N, M |
Insulinproduzierende Tumoren sind in über 90 % der Fälle gutartig, dagegen sind Gastrinome, Glukagonome, VIPome und ACTH-produzierende sowie die nichtfunktionellen PanNEN meistens bösartig. Sie wachsen und metastasieren vergleichsweise langsam, sodass auch Patienten mit Metastasen noch eine mittlere Überlebensdauer von über vier Jahren erreichen.
Symptome und Diagnostik

Das Leitsymptom des Pankreaskopfkarzinoms ist eine stetig zunehmende, nicht von Koliken begleitete Gelbsucht (Ikterus), die durch die Verengung des Gallengangs verursacht wird. Diese Gelbsucht ist nur bei Papillentumoren ein Frühsymptom, ansonsten Zeichen eines fortgeschrittenen Befundes. In den Rücken ausstrahlende Bauchschmerzen sind ebenfalls häufig, aber uncharakteristisch. Allerdings sind die quälenden, über Monate langsam zunehmenden dumpfen, nachts verschlimmerten Schmerzen, die durch Infiltration des Solarplexus entstehen, oft der erste Anlass, einen Arzt aufzusuchen. Ein Courvoisier-Zeichen (prall tastbare Gallenblase) ist möglich. Verengungen des Pankreasgangs beeinträchtigen die Drüsenfunktion und verursachen Verdauungsbeschwerden, Gewichtsverlust um mehr als 10 % und Diabetes. Thrombosen und neue Pigmentierungen der Haut sind Warnhinweise auf einen Tumor im Bauchraum. Im späten Erkrankungsstadium können Metastasen zur Lebervergrößerung, Leberfunktionsstörung, Bauchwassersucht und hochgradige Abmagerung führen.
Im Serum findet man unspezifische Entzündungsparameter wie CRP erhöht, außerdem die Pankreasenzyme Trypsin, Lipase und Amylase. Als Tumormarker werden CA 19-9 und mit Einschränkung auch CEA genannt, die jedoch nicht spezifisch (CA 19-9: Sensitivität und Spezifität circa 70 %) und als Screeningparameter deshalb ungeeignet sind. Bessere diagnostische Parameter werden noch gesucht; die Forschung konzentriert sich gegenwärtig auf Proteomik (Eiweißprofile), microRNAs und auf KRAS-Mutationen in Serum und Gallenflüssigkeit, wobei letzterer Parameter in Vorstudien über 90 % Sensitivität und Spezifität erreichte.[19]
Nichtinvasive Untersuchungsverfahren wie Sonografie, Computertomographie und Kernspintomographie stehen an erster Stelle der apparativen Diagnostik, erbringen aber nicht immer ein eindeutiges Ergebnis. Die ERCP (eine Kombination aus Endoskopie und Röntgenkontrastdarstellung) kann den Verschluss des Gallen- oder Pankreasganges nachweisen und bei günstiger Lage eine Bioptat des Tumors ermöglichen. Die Endosonografie funktioniert ähnlich, mit einer hohen Genauigkeit in der Beurteilung des Tumors und möglicher Metastasen in dessen Umgebung. Spezialkliniken halten gelegentlich auch die neu entwickelte Pankreatikoskopie vor, eine Endoskopie bis in das Pankreasgangsystem hinein, die noch in der Erprobung steht und die auf das Gangsystem beschränkte Neubildungen möglicherweise am besten darstellen kann.
Wenn die genannten Diagnosemethoden nicht ausreichen, um eine schwere chronische Pankreatitis sicher von einem Tumor unterscheiden zu können, oder um eine Peritonealkarzinose nachzuweisen, bleibt zuletzt noch die Bauchspiegelung (Laparoskopie). Dieser Eingriff wird heute meist mit einer laparoskopischen Ultraschallsonde und einer Bauchspülung kombiniert, was als „erweiterte diagnostische Laparoskopie“ (EDL) bezeichnet wird.
Die klinischen und apparativen Untersuchungen liefern die Tumordiagnose und das Tumorstadium (Grad der Ausbreitung). Die TNM-Klassifikation dient zur international einheitlichen Klassifikation der Ausbreitung bösartiger Tumoren. Beim Pankreaskarzinom wird sie wie folgt vorgenommen:
| T | Primärtumor |
|---|---|
| TX | Primärtumor kann nicht beurteilt werden |
| T0 | Kein Primärtumor nachweisbar |
| Tis | Carcinoma in situ (= frühestes, noch nicht invasives Tumorstadium) |
| T1 | Größter Durchmesser des Primärtumors ≤ 2 cm; Tumor noch innerhalb des Pankreas |
| T2 | Größter Durchmesser des Primärtumors > 2 cm; Tumor noch innerhalb des Pankreas |
| T3 | Tumor hat die Organgrenze überschritten, aber die Arterien noch nicht infiltriert |
| T4 | Angrenzende große Arterien sind infiltriert (Truncus coeliacus, A. mesenterica sup.) |
| N | Regionäre Lymphknoten |
| NX | Die regionären Lymphknoten können nicht beurteilt werden |
| N0 | Keine regionären Lymphknotenmetastasen |
| N1 | Regionäre Lymphknoten sind befallen |
| M | Fernmetastasen |
| MX | Fernmetastasen können nicht beurteilt werden |
| M0 | Keine Fernmetastasen |
| M1 | Fernmetastasen (einschließlich Lymphknoten am Pankreasschwanz) |
Aus der Ausbreitung ergibt sich das Tumorstadium, nach dem sich die Behandlung richten wird.
| Stadium | |
|---|---|
| IA | T1 N0 M0 (Tumor bis 2 cm, keine Metastasen) |
| IB | T2 N0 M0 (Tumor innerhalb des Pankreas, keine Metastasen) |
| IIA | T3 N0 M0 (Tumor noch operabel, keine Metastasen) |
| IIB | T1-3 N1 M0 (Tumor noch operabel, mit regionären Lymphknotenmetastasen) |
| III | T4 N0-1 M0 (Lokal fortgeschritten, ohne Fernmetastasen) |
| IV | T1-4 N0-1 M1 (Fernmetastasen) |
Behandlung

Gestaute Gallenwege können zunächst mit einem endoskopisch platzierten Stent (Röhrchen) freigemacht werden, um den Allgemeinzustand des Patienten zu verbessern. Es gibt auch Verfahren, bei denen die Galle aus der Leber durch einen Katheter nach außen abgeleitet wird. Ist beides unmöglich, kann ein begrenzter chirurgischer Eingriff Entlastung verschaffen. Bei operablen Tumoren wird wegen der Gefahr von aufsteigenden Infektionen und Wundheilungsstörungen in der Regel auf die vorherige Entlastung verzichtet, mit Ausnahme von schwersten Stauungen (z. B. Bilirubin-Spiegel > 10 mg/dl). Weitere Erstmaßnahmen richten sich gegen die oft (25 %) bestehende Mangelernährung, die das Operationsrisiko deutlich erhöht, gegen Eiweiß- und Vitaminmangel (insbesondere fettlösliche Vitamine) und gegen diabetische Stoffwechselstörungen.
Um die Versorgung zu verbessern und aktuellen Erkenntnissen gerecht zu werden, hat die Deutsche Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselerkrankungen (DGVS) im Dezember 2013 eine aktualisierte S3-Leitlinie „Exokrines Pankreaskarzinom“ herausgegeben.[21]
Operative Entfernung
Etwa vier von fünf Pankreaskarzinomen sind, wenn sie erkannt werden, schon zu weit fortgeschritten und können nicht mehr mit dem Ziel der Heilung (kurativ) operiert werden. Auch wenn nur einzelne Lebermetastasen vorliegen, kann man – im Unterschied zum Darmkrebs – mit deren Entfernung keine Heilung erreichen. Tumoren, die weder große Arterien infiltriert noch Fernmetastasen verursacht haben (UICC-Stadien I und II), können prinzipiell noch komplett entfernt werden. Infiltrierte Venen machen den Eingriff nicht unmöglich; befallene Lymphknoten werden mit entfernt. Ob auch nicht befallene Lymphknoten sicherheitshalber entfernt werden sollten, ist umstritten.
Es ist sinnvoll, möglichst einen Teil des Organs zu erhalten und wieder mit dem Darm zu verbinden. Je nach Ort des Tumors wird deshalb die rechtsseitige Teilresektion (Duodenopankreatektomie, mehrere Varianten), eine mittlere Teilresektion, eine linksseitige Teilresektion (Pankreasschwanzresektion) oder eine Totalresektion (vollständige Entfernung) der Bauchspeicheldrüse durchgeführt, meist einschließlich aller regionären Lymphknoten. Der Magen, der Gallengang und gegebenenfalls der Ausführungsgang des belassenen Rests der Bauchspeicheldrüse müssen wieder mit dem Darm verbunden werden. Man verwendet dafür eine oder mehrere hochgezogene Dünndarmschlingen, die spannungsfrei angeschlossen und untereinander verbunden werden, basierend auf der klassischen von César Roux erdachten Methode.
Alle genannten Eingriffe sind schwerwiegend und kompliziert. Frühe Komplikationen wie Pankreatitis, Sepsis, Anastomoseninsuffizienz (Lecks in den Darmnähten), Wundheilungsstörungen und Fisteln (abnorme Gangsysteme) sind sehr häufig. Wird der Hauptlymphgang verletzt, kann ein Chylaskos (Lymphaustritt in die Bauchhöhle) die Folge sein. Die bedrohlichste Komplikation ist die Nachblutung aus großen Blutgefäßen, die vor allem bei mehrfachen perioperativen Manipulationen (ERCP) oder bei Entzündungen im Operationsgebiet droht. Diabetes bekommen etwa 10 % der Patienten nach Teilresektionen und praktisch alle nach Totalresektionen des Pankreas. Die fehlenden Verdauungsenzyme müssen in der Folge lebenslang in Tablettenform ersetzt werden.
- Operationsvarianten
-
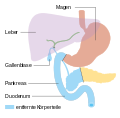 Pylorus-erhaltende Teilresektion
Pylorus-erhaltende Teilresektion -
 Duodenopankreatektomie nach Whipple
Duodenopankreatektomie nach Whipple -
 Endzustand nach Whipple-Operation
Endzustand nach Whipple-Operation -
 Totale Pankreasresektion
Totale Pankreasresektion -
 Endzustand nach Totalresektion
Endzustand nach Totalresektion





Die Enukleation (Ausschälung) ist eine sehr gewebesparende Technik, bei der nur der Tumor entfernt wird. Sie ist für gutartige Befunde und oberflächlich gelegene neuroendokrine Tumoren geeignet. Der Tumor muss Abstand vom Gangsystem haben.
Mit einer Segmentresektion werden definierte Teile der Bauchspeicheldrüse entfernt: entweder der Kopf einschließlich Zwölffingerdarm, der mittlere (zentrale) Abschnitt oder der linksseitige Teil. Die Milz kann mit entfernt oder bei geeigneten Fällen belassen werden. Vom Ausführungsgang abgeschnittenes Gewebe muss entweder in eine nach Roux Y-förmig (d. h. end-zu-seit-) angenähte Dünndarmschlinge oder in den Magen drainiert werden.
Die den pyloruserhaltende Duodenopankreatektomie nach den kalifornischen Chirurgen Longmire und Traverso[22] erhält im Gegensatz zu der Kausch-Whipple-Operation den gesamten Magen bis zum Pförtnermuskel (Pylorus).
Die subtotale oder totale Pankreasentfernung bringt als Maximaleingriff die höchste Letalität mit sich. In etwa 6 % der Operationen kommt sie zum Einsatz. Gefürchtete Komplikation ist der schwer einstellbare Brittle-Diabetes.
Inwieweit die benachbarten Lymphknoten vorbeugend ebenfalls zu entfernen sind, ist noch nicht abschließend geklärt. Jeder zweite Patient hat bei der Krankenhausaufnahme schon Lymphknotenmetastasen entwickelt. Die Standard-Lymphonodektomie umfasst die um die bauchspeicheldrüse und am Zwölffingerdarm liegenden Lymphknoten sowie jene rechts der oberen Gekrösearterie und teilweise der aus dem Leber-Zwölffingerdarm-Band. Das radikale Konzept skelettiert komplett den Truncus coeliacus, das Leber-Zwölffingerdarm-Band und die Aorta auf Höhe der Bauchspeicheldrüse. Ein noch weitergehendes japanisches Vorgehen[23] schließt sämtliche Lymphknoten an der Bauchaorta vom Zwerchfell bis zur Endaufzweigung ein. Bisher ist eine verbesserte Heilungsrate dieser Varianten nicht nachgewiesen.
Palliativoperationen dienen zur Symptomlinderung. Beispielsweise wird mit der Choledochojejunostomie eine Verbindung zwischen dem gestauten Gallengang und dem Darm geschaffen, um die Galle aus der Leber abzuleiten.
Chemo- und Strahlentherapie
Eine vor der Operation durchgeführte („neoadjuvante“) Chemotherapie kann nach der gegenwärtigen Phase-II-Studienlage einige fortgeschrittene Tumoren soweit verkleinern, dass sie operabel werden, die Daten sind aber noch zu schwach für eine allgemeine Empfehlung.[24] Gleiches gilt für die neoadjuvante Kombination aus Chemo- und Strahlentherapie (Radiochemotherapie), die in kleineren Studien erprobt wird. Nur wenn die Standardchemotherapie nicht mehr wirkt, nutzt man als second line eine Kombination aus Oxaliplatin, 5-Fluoruracil (5-FU) und Folinsäure (OFF-Schema). Als Erstlinientherapie gegen neuroendokrine Tumoren empfiehlt die ENET die Kombination von Streptozocin mit Doxorubicin oder 5-FU; bei hochgradig bösartigen neuroendokrinen Tumoren Cisplatin und Etoposid. Alternativ werden Temozolomid und Capecitabin erprobt.
Dagegen ist eine nach der Operation (adjuvante) durchgeführte Chemotherapie anerkannter Standard, weil die Rezidivrate (s. u.) extrem hoch ist.[25] Jede Woche 1x 1000 mg/(m² KOF) Gemcitabin drei Wochen lang, dann eine Woche Pause, dann der nächste Zyklus, ist das in Europa gebräuchlichste Schema. In den USA wird eher 5-Fluoruracil eingesetzt. Kombinationen mehrerer Zytostatika verbessern die Wirkung bei Karzinomen bisher nicht, sind allerdings der Standard bei NET. Radiochemotherapien erhöhen nachweislich die örtliche Tumorkontrolle, allerdings wird dabei weder Metastasierung noch Sterblichkeit verringert, sodass darauf meist verzichtet wird.
Nicht heilbare Tumoren können mit einer Chemotherapie palliativ behandelt werden, z. B. mit Gemcitabin.[26] Auch eine vorsichtig dosierte Radiochemotherapie kann zur Schmerzlinderung beitragen. Ein anderer Ansatz ist es, das Zytostatikum über einen von der Leistenarterie eingebrachten Katheter in eine tumorversorgende Arterie zu spritzen, z. B. in die Arteria pancreatica magna (lokoregionäre Chemotherapie). Damit kann die Dosis am Tumor ohne zusätzliche Nebenwirkungen gesteigert werden. Es gibt aber bisher nur Phase-I-Studien (kleine Fallserien) zu dieser teuren und technisch sehr aufwändigen Methode.
Wie bei vielen anderen Krebserkrankungen setzt die Medizin auch beim Pankreaskarzinom zunehmend auf die Gezielte Krebstherapie (targeted therapies), d. h. auf monoklonale Antikörper, andere Biologika und small molecules. Außerhalb von Studien ist der Tyrosinkinase-Inhibitor Erlotinib gegen Pankreaskarzinome zugelassen; außerdem der Tyrosinkinase-Inhibitor Sunitinib und der mTOR-Inhibitor Everolimus gegen endokrine Tumoren. Diese Substanzen haben eine hohe Ansprechrate – einzelne Patienten blieben jahrelang stabil –, aber auch ein erhebliches Nebenwirkungspotential.
Gegen die durch hormonproduzierende (funktionell aktive) Tumoren verursachten Symptome können Somastostatinanaloga wie Octreotid und Interferon-alpha eingesetzt werden, die Krankheit wird damit allerdings nicht aufgehalten. Beim Versagen der Erstlinien-Chemotherapie kann die Peptid-Rezeptor-Radionuklidtherapie (PRRT) versucht werden, bei der ein wirksames Radionuklid chemisch an einen geeigneten Liganden gekoppelt in die Blutbahn injiziert wird.
Prognose

Mit den verbesserten Operationstechniken und der Chemotherapie sind die Heilungsraten etwas besser geworden. Die Prognose der Pankreaskarzinome ist jedoch immer noch eine der schlechtesten aller Karzinome: Neueren Untersuchungen zufolge liegt die Fünf-Jahres-Überlebensrate nach einem in kurativer Absicht durchgeführten Eingriff mit anschließender Chemotherapie bei 20 %,[27] höchstens 30 %.[28] Leider sind nur 10 bis 20 % der Tumoren zum Zeitpunkt der Diagnose noch operabel. Bei vier von fünf operierten Patienten kehrt der Tumor innerhalb von zwei Jahren zurück und nur in vereinzelten Fällen kann er ein zweites Mal entfernt werden. Inoperable Tumoren sprechen nur begrenzt auf eine Chemotherapie an; diese Patienten haben nur eine mittlere Überlebensdauer von vier bis sieben Monaten, die sich auch mit palliativer Chemotherapie nicht wesentlich verlängern lässt.[25]
In dieser Situation liegt es in der Verantwortung der Onkologen, ihre therapeutischen Entscheidungen unter das Primat der subjektiven Lebensqualität des Patienten zu stellen. Dieser hermeneutische Endpunkt muss neben den mechanistischen Endpunkten wie Überlebensrate, Überlebenszeit, progressionsfreies Überleben etc. mindestens gleichwertig berücksichtigt werden. Das ärztliche Handeln sollte sich nach standardisierten Behandlungspfaden (clinical pathways) richten und sowohl die wissenschaftliche Evidenz als auch die wirtschaftliche Effizienz berücksichtigen.
Für die Lebensqualität wichtige Faktoren sind eine adäquate, ausreichend hoch dosierte Schmerztherapie nach dem WHO-Stufenschema und fachkundige Hilfen zur psychologischen Krankheitsbewältigung. Best Supportive Care bedeutet, sich von unheilbaren Patienten nicht zurückzuziehen, sondern ihnen im Gegenteil jederzeit und dauerhaft medizinische Hilfe anzubieten. Beispielsweise ist es für Krebspatienten wichtig, sich möglichst lange normal ernähren zu können; dafür sind geeignete Diätprodukte zu verordnen und mechanische Hindernisse soweit möglich, auch mit palliativen Eingriffen, zu beheben. In der Endphase können Stents in den Darmtrakt eingesetzt werden, die zumindest für Brei- und Flüssignahrung passierbar sind, oder auf Sondenkost über eine PEG umgestellt werden. Neben dem Tumorschmerz ist Juckreiz ein weiteres sehr belastendes Symptom des fortgeschrittenen Pankreaskarzinoms, das in einem sorgfältigen Palliativkonzept durchaus gezielt und wirksam behandelt werden kann.
Darüber hinaus benötigen auch geheilte Patienten Betreuung. Wesentliche Ziele ihrer Rehabilitation sind Kostaufbau und Ernährungsberatung, Enzymsubstitution, Diabeteskontrolle und psychotherapeutische Stützungsmaßnahmen wie Gruppen- und Einzelgespräche. Sozialmedizinische Hilfen durch entsprechendes Fachpersonal sind Beratungen nach dem Schwerbehindertenrecht, Leistungen der beruflichen Rehabilitation, Häusliche Krankenpflege, Haushaltshilfe, Essensversorgung usw. Sie müssen noch im Krankenhaus im Rahmen eines geplanten Entlassungsmanagements eingeleitet werden.
Quellen
- H. U. Baer, M. Wagner, M. W. Büchler: Onkologische Standardchirurgie des Pankreaskarzinoms. In: Chir Gastroenterol. Band 14, 1998, S. 42–48.
- Hans G. Beger, Markus W. Büchler, Henning Dralle, Markus M. Lerch, Peter Malfertheiner, Joachim Mössner, Jürgen F. Riemann: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3642379648
- M. Birth, Thomas Heinz Ittel, Philippe L. Pereira: Hepatobiliäre und Pankreastumoren. Springer-Verlag, 2010, ISBN 3-642-04935-4
- F. T. Bosman, F. Carneiro, R. H. Hruban, N. D. Theise: WHO Classification of Tumours of the Digestive System. 4. Auflage, Band 3 der Reihe WHO Classification of Tumours. WHO/IARC 2010. ISBN 978-9-283-22432-7
- R. Grützmann u. a.: Intraduktale papillär-muzinöse Neoplasie des Pankreas: Aktueller Stand von Diagnostik, Therapie und Prognose. In: Dtsch Arztebl Int. Nr. 108(46), 2011, S. 788–794 (Übersichtsarbeit).
Weiterführende Literatur
- T. Seufferlein, J. B. Bachet u. a.: Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. In: Annals of oncology. Band 23 Suppl 7, Oktober 2012, ISSN 1569-8041, S. vii33–vii40, doi:10.1093/annonc/mds224. PMID 22997452.
Weblinks
- S3-Leitlinie Exokrines Pankreaskarzinom, AWMF-Registernummer 032/010, Stand 10/2013
- Informationsseite und "blauer Ratgeber" (Patientenbroschüre, Stand 12/2012, PDF) der Deutschen Krebsgesellschaft
- Kampagne „Aus der Mitte - Diagnose Bauchspeicheldrüsenkrebs“ von Hoffmann-La Roche
- Selbsthilfeverein Tumore und Erkrankungen der Bauchspeicheldrüse e. V.
- Pancreatic Cancer. NCCN Guidelines for patients. National Comprehensive Cancer Network, Jan. 2014 (PDF, in englischer Sprache)
- Pancreatica:Confronting pancreatic cancer. Webangebot in englische Sprache, von der Cancer Patients Alliance (US-amerikanische Stiftung)
- Liste der DGAV-zertifizierten Zentren für Pankreaschirurgie in Deutschland
- Häufige Krankheiten – modern behandelt: Bauchspeicheldrüsenkrebs. In: nzz.ch. 21. Februar 2013, abgerufen am 17. Januar 2015.
Einzelnachweise
- ↑ John Malone Howard und Walter Hess: History of the Pancreas: Mysteries of a Hidden Organ. Springer, 2002, ISBN 978-0-306-46742-4, S. 6.
- ↑ John Malone Howard und Walter Hess: History of the Pancreas: Mysteries of a Hidden Organ. Springer 2002, ISBN 978-0-306-46742-4, S. 83.
- ↑ John Malone Howard und Walter Hess: History of the Pancreas: Mysteries of a Hidden Organ. Springer, 2002, ISBN 978-0-306-46742-4, S. 107.
- ↑ John Malone Howard und Walter Hess: History of the Pancreas: Mysteries of a Hidden Organ. Springer, 2002, ISBN 978-0-306-46742-4, S. 117.
- ↑ Christos G. Dervenis, Claudio Bassi: Pancreatic Tumors: Achievements and Perspectives. Thieme, 2000, ISBN 978-1-58890-002-9, S. 4 (eingeschränkte Vorschau in der Google-Buchsuche).
- ↑ Detlev Drenckhahn (Hrsg.): Anatomie, Band 1. 17. Auflage. Urban&Fischer, München 2008, ISBN 978-3-437-42342-0, S. 723.
- ↑ Nicolas T. Schwarz, Karl-Heinz Reutter: Allgemein- und Viszeralchirurgie essentials: Intensivkurs zur Weiterbildung. 7. Auflage. Georg Thieme, 2012, ISBN 978-3-13-159057-2, S. 256.
- ↑ F. G. Bader u. a.: Histopathologie, Tumorklassifikation und Prognosefaktoren. In: M. Birth, T. H. Ittel, P. L. Pereira: Hepatobiliäre und Pankreastumoren. Springer-Verlag, 2010, ISBN 3-642-04935-4, S. 91.
- ↑ Informationen zum Pankreastumor vom Zentrum für Krebsregisterdaten am Robert Koch-Institut Krebs - Bauchspeicheldrüsenkrebs. In: krebsdaten.de. 13. Dezember 2014, abgerufen am 16. Januar 2015.
- ↑ Zentrum für Krebsregisterdaten am Robert Koch-Institut und der Gesellschaft der epidemiologischen Krebsregister in Deutschland: Krebs - Krebs in Deutschland. In: krebsdaten.de. 13. Dezember 2013, abgerufen am 16. Januar 2015.
- ↑ N. Schulte: Epidemiologie und Karzinogenese des Pankreaskarzinoms. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 334. doi:10.1007/978-3-642-37964-2_62
- ↑ P. Langer, D. K. Bartsch: Familiäres Pankreaskarzinom. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 342–343. doi:10.1007/978-3-642-37964-2_63
- ↑ G. Andreotti, D. T. Silverman: Occupational risk factors and pancreatic cancer: a review of recent findings. In: Molecular Carcinogenesis. Band 51, Nummer 1, Januar 2012, S. 98–108, ISSN 1098-2744. doi:10.1002/mc.20779. PMID 22162234. (Review).
- ↑ A. F. Hezel, A. C. Kimmelman, B. Z. Stanger, N. Bardeesy, R. A. Depinho: Genetics and biology of pancreatic ductal adenocarcinoma. In: Genes Dev. 20. Jahrgang, Nr. 10, Mai 2006, S. 1218–1249, doi:10.1101/gad.1415606, PMID 16702400 (cshlp.org).
- ↑ C. Wittekind: Pathohistologische Klassifikation, Tumorstaging und R-Klassifikation des Pankreaskarzinoms. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 327. doi:10.1007/978-3-642-37964-2_61
- ↑ R. H. Hruban, G. Klöppel, P. Bofetta u. a.: Tumours of the pancreas. In: F. T. Bosman u. a.: WHO Classification of Tumours of the Digestive System. 4. Auflage, Band 3 der Reihe WHO Classification of Tumours. WHO/IARC 2010. ISBN 978-9-283-22432-7, S. 279–337.
- ↑ G. Klöppel: Klassifikation und Pathologie endokriner Tumoren des Pankreas. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 204–209. doi:10.1007/978-3-642-37964-2_39
- ↑ K. Streetz, W. Karges: Laborchemische und genetische Diagnostik endokriner Tumoren des Pankreas. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 216–217. doi:10.1007/978-3-642-37964-2_41
- ↑ J. M. Winter, C. J. Yeo, J. R. Brody: Diagnostic, prognostic, and predictive biomarkers in pancreatic cancer. In: Journal of surgical oncology. Band 107, Nummer 1, Januar 2013, S. 15–22, ISSN 1096-9098. doi:10.1002/jso.23192. PMID 22729569. (Review).
- ↑ Exocrine and endocrine pancreas. In: S. B. Edge, D. R. Byrd, C. C. Compton u. a. (Hrsg.): AJCC Cancer Staging Manual. 7. Auflage, New York, NY: Springer, 2010, S. 241–249.
- ↑ DGVS: Leitlinie Exokrines Pankreaskarzinom
- ↑ W. P. Longmire, L. W. Traverso: The Whipple procedure and other standard operative approaches to pancreatic cancer. In: Cancer. Band 47, Nummer 6 Suppl, März 1981, ISSN 0008-543X, S. 1706–1711, PMID 6791804.
- ↑ O. Ishikawa, H. Ohhigashi u. a.: Practical usefulness of lymphatic and connective tissue clearance for the carcinoma of the pancreas head. In: Annals of Surgery. Band 208, Nummer 2, August 1988, S. 215–220, ISSN 0003-4932. PMID 2840866. PMC 1493620 (freier Volltext).
- ↑ H. Oettle, M. Sinn: Chemotherapie beim Pankreaskarzinom. In: M. Birth, T. H. Ittel, P. L. Pereira: Hepatobiliäre und Pankreastumoren. Springer-Verlag, 2010, ISBN 3-642-04935-4, S. 381.
- ↑ a b Heinemann V: Evidenz der Chemotherapie beim fortgeschrittenen Pankreaskarzinom. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 377–382. doi:10.1007/978-3-642-37964-2_71
- ↑ H. A. Burris, M. J. Moore u. a.: Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. In: Journal of clinical oncology : official journal of the American Society of Clinical Oncology. Band 15, Nummer 6, Juni 1997, ISSN 0732-183X, S. 2403–2413, PMID 9196156.
- ↑ O. Strobel, J. Werner: Langzeitverlauf nach operativer Tumorentfernung und Chemotherapie des duktalen Pankreaskarzinoms. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 415–418. doi:10.1007/978-3-642-37964-2_79
- ↑ Wagner 2004 und Schmidt 2012, zitiert nach: T. Hackert, W. Hartwig: Indikation zur Resektion beim Pankreaskarzinom. In: H. G. Beger u. a.: Erkrankungen des Pankreas: Evidenz in Diagnostik, Therapie und Langzeitverlauf. Springer-Verlag, 2013. ISBN 3-642-37964-8, S. 365. doi:10.1007/978-3-642-37964-2_68